Porphyria is a group of disorders in which substances called porphyrins build up in the body, adversely affecting the skin or nervous system. The types that affect the nervous system are also known as acute porphyria, as symptoms are rapid in onset and short in duration. Symptoms of an attack include abdominal pain, chest pain, vomiting, confusion, constipation, fever, high blood pressure, and high heart rate. The attacks usually last for days to weeks. Complications may include paralysis, low blood sodium levels, and seizures. Attacks may be triggered by alcohol, smoking, hormonal changes, fasting, stress, or certain medications. If the skin is affected, blisters or itching may occur with sunlight exposure.

Heme, or haem, is a ring-shaped iron-containing molecular component of hemoglobin, which is necessary to bind oxygen in the bloodstream. It is composed of four pyrrole rings with 2 vinyl and 2 propionic acid side chains. Heme is biosynthesized in both the bone marrow and the liver.

In the fields of biochemistry and pharmacology an allosteric regulator is a substance that binds to a site on an enzyme or receptor distinct from the active site, resulting in a conformational change that alters the protein's activity, either enhancing or inhibiting its function. In contrast, substances that bind directly to an enzyme's active site or the binding site of the endogenous ligand of a receptor are called orthosteric regulators or modulators.
Phenylalanine hydroxylase (PAH) (EC 1.14.16.1) is an enzyme that catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. PAH is one of three members of the biopterin-dependent aromatic amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin (BH4, a pteridine cofactor) and a non-heme iron for catalysis. During the reaction, molecular oxygen is heterolytically cleaved with sequential incorporation of one oxygen atom into BH4 and phenylalanine substrate. In humans, mutations in its encoding gene, PAH, can lead to the metabolic disorder phenylketonuria.
Aminolevulinic acid synthase (ALA synthase, ALAS, or delta-aminolevulinic acid synthase) is an enzyme (EC 2.3.1.37) that catalyzes the synthesis of δ-aminolevulinic acid (ALA) the first common precursor in the biosynthesis of all tetrapyrroles such as hemes, cobalamins and chlorophylls. The reaction is as follows:
Dehydratases are a group of lyase enzymes that form double and triple bonds in a substrate through the removal of water. They can be found in many places including the mitochondria, peroxisome and cytosol. There are more than 150 different dehydratase enzymes that are classified into four groups. Dehydratases can act on hydroxyacyl-CoA with or without cofactors, and some have a metal and non-metal cluster act as their active site.
Acute intermittent porphyria (AIP) is a rare metabolic disorder affecting the production of heme resulting from a deficiency of the enzyme porphobilinogen deaminase. It is the most common of the acute porphyrias.

Protoporphyrinogen oxidase or protox is an enzyme that in humans is encoded by the PPOX gene.
Porphobilinogen deaminase (hydroxymethylbilane synthase, or uroporphyrinogen I synthase) is an enzyme (EC 2.5.1.61) that in humans is encoded by the HMBS gene. Porphobilinogen deaminase is involved in the third step of the heme biosynthetic pathway. It catalyzes the head to tail condensation of four porphobilinogen molecules into the linear hydroxymethylbilane while releasing four ammonia molecules:
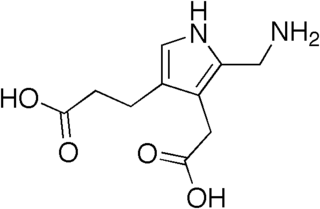
Porphobilinogen (PBG) is an organic compound that occurs in living organisms as an intermediate in the biosynthesis of porphyrins, which include critical substances like hemoglobin and chlorophyll.
Erythropoietic porphyria is a type of porphyria associated with erythropoietic cells. In erythropoietic porphyrias, the enzyme deficiency occurs in the red blood cells.

Aspartate kinase or aspartokinase (AK) is an enzyme that catalyzes the phosphorylation of the amino acid aspartate. This reaction is the first step in the biosynthesis of three other amino acids: methionine, lysine, and threonine, known as the "aspartate family". Aspartokinases are present only in microorganisms and plants, but not in animals, which must obtain aspartate-family amino acids from their diet. Consequently, methionine, lysine and threonine are essential amino acids in animals.

Cystathionine-β-synthase, also known as CBS, is an enzyme (EC 4.2.1.22) that in humans is encoded by the CBS gene. It catalyzes the first step of the transsulfuration pathway, from homocysteine to cystathionine:

D-bifunctional protein (DBP), also known as peroxisomal multifunctional enzyme type 2 (MFP-2), as well as 17β-hydroxysteroid dehydrogenase type IV is a protein that in humans is encoded by the HSD17B4 gene. It's an alcohol oxidoreductase, specifically 17β-Hydroxysteroid dehydrogenase. It is involved in fatty acid β-oxidation and steroid metabolism.

Pterin-4-alpha-carbinolamine dehydratase is an enzyme that in humans is encoded by the PCBD1 gene.

Delta-aminolevulinate synthase 2 also known as ALAS2 is a protein that in humans is encoded by the ALAS2 gene. ALAS2 is an aminolevulinic acid synthase.

Delta-aminolevulinate synthase 1 also known as ALAS1 is a protein that in humans is encoded by the ALAS1 gene. ALAS1 is an aminolevulinic acid synthase.

Morpheeins are proteins that can form two or more different homo-oligomers, but must come apart and change shape to convert between forms. The alternate shape may reassemble to a different oligomer. The shape of the subunit dictates which oligomer is formed. Each oligomer has a finite number of subunits (stoichiometry). Morpheeins can interconvert between forms under physiological conditions and can exist as an equilibrium of different oligomers. These oligomers are physiologically relevant and are not misfolded protein; this distinguishes morpheeins from prions and amyloid. The different oligomers have distinct functionality. Interconversion of morpheein forms can be a structural basis for allosteric regulation, an idea noted many years ago, and later revived. A mutation that shifts the normal equilibrium of morpheein forms can serve as the basis for a conformational disease. Features of morpheeins can be exploited for drug discovery. The dice image represents a morpheein equilibrium containing two different monomeric shapes that dictate assembly to a tetramer or a pentamer. The one protein that is established to function as a morpheein is porphobilinogen synthase, though there are suggestions throughout the literature that other proteins may function as morpheeins.

Robert J. Desnick is an American human geneticist whose basic and translational research accomplishments include significant discoveries in genomics, pharmacogenetics, gene therapy, personalized medicine, and the treatment of genetic diseases. His translational research has led to the development of the enzyme replacement therapy (ERT) and the chaperone therapy for Fabry disease, ERT for Niemann–Pick disease type B, and the RNA Interference Therapy for the Acute Hepatic Porphyrias.

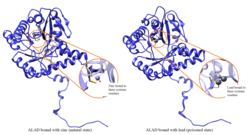
Aminolevulinic acid dehydratase deficiency porphyria is an extremely rare autosomal recessive metabolic disorder that results from inappropriately low levels of the enzyme delta-aminolevulinic acid dehydratase (ALAD), which is required for normal heme synthesis. This deficiency results in the accumulation of a toxic metabolic precursor in the heme synthesis pathway called aminolevulinic acid (ALA). Lead poisoning can also disrupt ALAD and result in elevated ALA causing the same symptoms. Heme is a component of hemoglobin which carries oxygen in red blood cells.