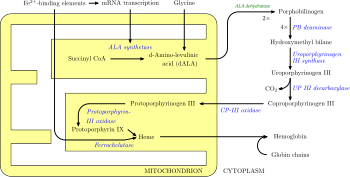
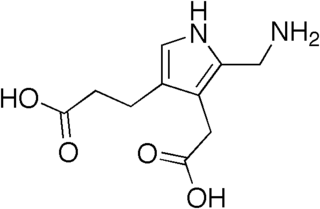
Heme, or haem, is a ring-shaped iron-containing molecular component of hemoglobin, which is necessary to bind oxygen in the bloodstream. It is composed of four pyrrole rings with 2 vinyl and 2 propionic acid side chains. Heme is biosynthesized in both the bone marrow and the liver.

Hereditary coproporphyria (HCP) is a disorder of heme biosynthesis, classified as an acute hepatic porphyria. HCP is caused by a deficiency of the enzyme coproporphyrinogen oxidase, coded for by the CPOX gene, and is inherited in an autosomal dominant fashion, although homozygous individuals have been identified. Unlike acute intermittent porphyria, individuals with HCP can present with cutaneous findings similar to those found in porphyria cutanea tarda in addition to the acute attacks of abdominal pain, vomiting and neurological dysfunction characteristic of acute porphyrias. Like other porphyrias, attacks of HCP can be induced by certain drugs, environmental stressors or diet changes. Biochemical and molecular testing can be used to narrow down the diagnosis of a porphyria and identify the specific genetic defect. Overall, porphyrias are rare diseases. The combined incidence for all forms of the disease has been estimated at 1:20,000. The exact incidence of HCP is difficult to determine, due to its reduced penetrance.

Variegate porphyria, also known by several other names, is an autosomal dominant porphyria that can have acute symptoms along with symptoms that affect the skin. The disorder results from low levels of the enzyme responsible for the seventh step in heme production. Heme is a vital molecule for all of the body's organs. It is a component of hemoglobin, the molecule that carries oxygen in the blood.

Porphyria cutanea tarda is the most common subtype of porphyria. The disease is named because it is a porphyria that often presents with skin manifestations later in life. The disorder results from low levels of the enzyme responsible for the fifth step in heme production. Heme is a vital molecule for all of the body's organs. It is a component of hemoglobin, the molecule that carries oxygen in the blood.

Erythropoietic protoporphyria is a form of porphyria, which varies in severity and can be very painful. It arises from a deficiency in the enzyme ferrochelatase, leading to abnormally high levels of protoporphyrin in the red blood cells (erythrocytes), plasma, skin, and liver. The severity varies significantly from individual to individual.

Gunther disease is a congenital form of erythropoietic porphyria. The word porphyria originated from the Greek word porphura. Porphura actually means "purple pigment", which, in suggestion, the color that the body fluid changes when a person has Gunther's disease. It is a rare, autosomal recessive metabolic disorder affecting heme, caused by deficiency of the enzyme uroporphyrinogen cosynthetase. It is extremely rare, with a prevalence estimated at 1 in 1,000,000 or less. There have been times that prior to birth of a fetus, Gunther's disease has been shown to lead to anemia. In milder cases patients have not presented any symptoms until they have reached adulthood. In Gunther's disease, porphyrins are accumulated in the teeth and bones and an increased amount are seen in the plasma, bone marrow, feces, red blood cells, and urine.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are often referred to as congenital metabolic diseases or inherited metabolic disorders. Another term used to describe these disorders is "enzymopathies". This term was created following the study of biodynamic enzymology, a science based on the study of the enzymes and their products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene-one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.
Acute intermittent porphyria (AIP) is a rare metabolic disorder affecting the production of heme resulting from a deficiency of the enzyme porphobilinogen deaminase. It is the most common of the acute porphyrias.

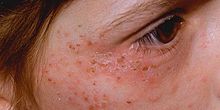
Polymorphous light eruption (PLE) presents with itchy red small bumps on sun-exposed skin, particularly face, neck, forearms and legs. It generally appears 30 minutes to a few hours after sun exposure and may last between one and 14 days. The bumps may become small blisters or plaques and may appear bloody,often healing with minimal scarring.

Uroporphyrinogen III decarboxylase is an enzyme that in humans is encoded by the UROD gene.

Heliophobia is the fear of the Sun, sunlight, or any bright light. It is a type of specific phobia.

Uroporphyrinogen III synthase is an enzyme involved in the metabolism of the cyclic tetrapyrrole compound porphyrin. It is involved in the conversion of hydroxymethyl bilane into uroporphyrinogen III. This enzyme catalyses the inversion of the final pyrrole unit of the linear tetrapyrrole molecule, linking it to the first pyrrole unit, thereby generating a large macrocyclic structure, uroporphyrinogen III. The enzyme folds into two alpha/beta domains connected by a beta-ladder, the active site being located between the two domains.

Protoporphyrin ferrochelatase (EC 4.98.1.1, formerly EC 4.99.1.1, or ferrochelatase; systematic name protoheme ferro-lyase (protoporphyrin-forming)) is an enzyme encoded by the FECH gene in humans. Ferrochelatase catalyses the eighth and terminal step in the biosynthesis of heme, converting protoporphyrin IX into heme B. It catalyses the reaction:
Hepatic porphyrias is a form of porphyria in which toxic porphyrin molecules build up in the liver. Hepatic porphyrias can result from a number of different enzyme deficiencies.
Erythropoietic porphyria is a type of porphyria associated with erythropoietic cells. In erythropoietic porphyrias, the enzyme deficiency occurs in the red blood cells.

Uroporphyrinogen I is an isomer of uroporphyrinogen III, a metabolic intermediate in the biosynthesis of heme. A type of porphyria is caused by production of uroporphyrinogen I instead of III.

Pseudoporphyria is a bullous photosensitivity that clinically and histologically mimics porphyria cutanea tarda. The difference is that no abnormalities in urine or serum porphyrin is noted on laboratories. Pseudoporphyria has been reported in patients with chronic kidney failure treated with hemodialysis and in those with excessive exposure to ultraviolet A (UV-A) by tanning beds.

Aminolevulinic acid dehydratase deficiency porphyria is an extremely rare autosomal recessive metabolic disorder that results from inappropriately low levels of the enzyme delta-aminolevulinic acid dehydratase (ALAD), which is required for normal heme synthesis. This deficiency results in the accumulation of a toxic metabolic precursor in the heme synthesis pathway called aminolevulinic acid (ALA). Lead poisoning can also disrupt ALAD and result in elevated ALA causing the same symptoms. Heme is a component of hemoglobin which carries oxygen in red blood cells.
Harderoporphyria is a rare disorder of heme biosynthesis, inherited in an autosomal recessive manner caused by specific mutations in the CPOX gene. Mutations in CPOX usually cause hereditary coproporphyria, an acute hepatic porphyria, however the K404E mutation in a homozygous or compound heterozygous state with a null allele cause the more severe harderoporphyria. Harderoporphyria is the first known metabolic disorder where the disease phenotype depended on the type and location of the mutations in a gene associated with multiple disorders.