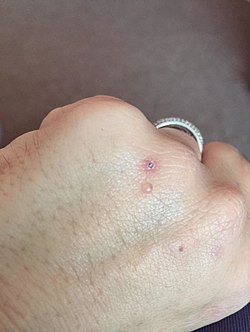
Porphyria cutanea tarda (PCT) is a form of long-term porphyria characterised by fragile skin and sore blisters in areas of skin that receive higher levels of exposure to sunlight, such as the face and backs of the hands.[1] These blisters burst easily resulting in erosions, crusts, and superficial ulcers.[1][2] There is often associated darkened skin color and extra facial hair growth.[1] Healing is typically slow, leading to scarring and milia, while changes such as hair loss, and alterations in nails may also occur.[1] A slightly purplish tint may be seen around the eyes.[1]Scleroderma-like thick skin may develop over fingers, scalp, behind the ears, at the back of the neck, or in the front of the chest.[1][2] The urine may appear dark.[2] Unlike other porphyrias, PCT does not cause severe illness.[2]
Tests generally include blood tests; liver function test, kidney function, complete blood count, ferritin, hepatitis B, hepatitis C, HIV, and HbA1C.[1][3] Other tests may include checking urine for pinky-red fluorescence with Wood's lamp, testing a 24-hour urine for porphyrins, and skin biopsy.[1][2]Pseudoporphyria may appear similar.[1] PCT may be distinguished from epidermolysis bullosa acquisita by the presence of excess hair.[3] Treatment for PCT focuses on removing all contributing factors, including alcohol, specific medicines, and managing hepatitis C and metabolic syndrome if applicable.[1] Though typical sunscreens may provide limited protection, more effective alternatives are barrier sunscreens.[1] Wearing a hat and gloves can help.[1] Other treatment options include phlebotomy.[3] An alternative is a low dose of an anti-malarial treatment.[3] Treating co-existing hepatitis C generally helps.[1] Without removing triggering factors, PCT tends to recur.[2] Recurrence in the year following phlebotomy treatment is not common.[3] Individuals with PCT have a 3.5 times higher risk of developing hepatocellular carcinoma.[1]
PCT is rare, though it is the most common subtype of porphyria.[1][4]
Signs and symptoms
PCT is typically characterised by fragile skin and sore blisters in areas of skin that receive higher levels of exposure to sunlight, such as the face and backs of the hands.[1] These blisters burst easily resulting in erosions, crusts, and superficial ulcers.[1][2] There is often associated darkened skin color and extra facial hair growth.[1] Healing is typically slow, leading to scarring and milia, while changes such as hair loss, and alterations in nails may also occur.[1] A slightly purplish tint may be seen around the eyes.[1]Scleroderma-like thick skin may develop over fingers, scalp, behind the ears, at the back of the neck, or in the front of the chest.[1][2] The urine may appear dark with a hint of reddishness.[2]
In addition to the skin lesions, chronic liver disease is very common in patients with sporadic PCT. This involves hepatic fibrosis (scarring of the liver), and inflammation. However, liver problems are less common in patients with the inherited form of the disease.[5] Additionally, patients will often void a wine-red color urine with an increased concentration of uroporphyrin I due to their enzymatic deficiency.[6]
Vitamin, mineral, and enzyme deficiencies
Certain vitamin and mineral deficiencies are common in people with porphyria cutanea tarda. The most common deficiencies are beta-Carotene,[7] retinol,[8] vitamin A[9] and vitamin C. Beta-Carotene is required to synthesize vitamin A, and vitamin A is needed to synthesize retinol. A lack of retinol-binding protein is due to a lack of retinol, which is required to trigger its production.[9]
Porphyrins interact with iron, absorbing photons to create reactive oxygen species is the mechanism of action causing the itchy, painful blisters of PCT.[7] The reactive oxygen species consume the skin antioxidants beta-carotene, vitamin E, and vitamin C. Supplementation of these three vitamins reduces the oxidation and potentially diminishes the severity of blister formation.[10] No single one of the three vitamins can inhibit the damaging effects of oxidized porphyrins, specifically uroporphyrins and coproporphyrins, but all three working together synergistically are capable of neutralizing their damaging effects.[citation needed]
Genetics
The reaction catalyzed by UroD20% of cases of porphyria cutanea tarda are inherited in an autosomal dominant pattern.
Inherited mutations in the URODgene cause about 20% of cases (the other 80% of cases do not have mutations in UROD, and are classified as sporadic). UROD makes an enzyme called uroporphyrinogen III decarboxylase, which is critical to the chemical process that leads to heme production. The activity of this enzyme is usually reduced by 50% in all tissues in people with the inherited form of the condition.[citation needed]
Nongenetic factors such as excess iron or partially genetic factors such as alcohol use disorder and others listed above can increase the demand for heme and the enzymes required to make heme. The combination of this increased demand and reduced activity of uroporphyrinogen decarboxylase disrupts heme production and allows byproducts of the process to accumulate in the body, triggering the signs and symptoms of porphyria cutanea tarda.[citation needed]
The HFE gene makes a protein that helps cells regulate the absorption of iron from the digestive tract and into the cells of the body. Certain mutations in the HFE gene cause hemochromatosis (an iron overload disorder). People who have these mutations are also at an increased risk of developing porphyria cutanea tarda.[citation needed]
In 20% of cases where porphyria cutanea tarda is inherited, it is inherited in an autosomal dominant pattern, which means one copy of the altered gene is sufficient to decrease enzyme activity and cause the signs and symptoms of the disorder.[citation needed]
Other
While inherited deficiencies in uroporphyrinogen decarboxylase often lead to the development of PCT, several risk factors can both cause and exacerbate the symptoms of this disease. One of the most common risk factors observed is infection with the Hepatitis C virus.[11] One review of a collection of PCT studies noted Hepatitis C infection in 50% of documented cases of PCT. Additional risk factors include alcohol use disorder, excess iron (from iron supplements as well as cooking on cast iron skillets), and exposure to chlorinated cyclic hydrocarbons and Agent Orange.[citation needed]
Porphyria cutanea tarda is primarily caused by uroporphyrinogen decarboxylase deficiency (UROD). Uroporphyrinogen decarboxylase occurs in nature as a homodimer of two subunits. It participates in the fifth step in the heme synthesis pathway, and is active in the cytosol. This enzymatic conversion results in coproporphyrinogen III as the primary product. This is accomplished by the clockwise removal of the four carboxyl groups present in the cyclic uroporphyrinogen III molecule. Therefore, a deficiency in this enzyme causes the aforementioned buildup of uroporphyrinogen and heptacarboxylic porphyrinogen, and to a lesser extent, hexacarboxylic porphyrinogen and pentacarboxylic porphyrinogen in the urine, which can be helpful in the diagnosis of this disorder.[16][17]
The dermatological symptoms of PCT, which include blistering and lesions on sun-exposed areas of the skin, are caused by a buildup of porphyrin compounds (specifically uroporphyrinogen) close to the surface of the skin that have been oxidized by free radicals or sunlight.[18] The oxidized porphyrins initiate degranulation of dermal mast cells,[19] which release proteases that catabolize the surrounding proteins.[20] This begins a cell-mediated positive feedback loop which matches the description of a type 4 delayed hypersensitivity reaction.[citation needed] The resulting blisters, therefore, do not appear immediately but begin to show up 2–3 days after sun exposure. Due to the highly conjugated structure of porphyrins involving alternating single and double carbon bonds, these compounds exhibit a deep purple color, resulting in the discoloration observed in the skin. Excess alcohol intake decreases hepcidin production, which leads to increased iron absorption from the gut and an increase in oxidative stress. This oxidative stress then leads to inhibition of uroporphyrinogen decarboxylase, creating an excess of uroporphyrinogen III which is oxidized from the relatively harmless porphyrinogens into their oxidized porphyrin form.[21] Concentrated instances of oxidative stress (alcohol, physical trauma, psychological stress, etc.) cause the liver to hemorrhage these porphyrins into the blood stream where they are then susceptible to oxidation.[citation needed]
The strong association of PCT with Hepatitis C virus infection is not entirely understood. Studies have suggested that the cytopathic effect of the virus on hepatocytes can lead to the release of free iron. This iron can disrupt the activity of cytochrome P450, releasing activated oxygen species. These can oxidize the UROD substrate uroporphyrinogen, which can result in the inhibition of UROD and lead to deficient activity of this key enzyme.[22]
Excess alcohol use is frequently associated with both inducing PCT[23] and aggravating a preexisting diagnosis of the disorder. It is thought to do so by causing oxidative damage to liver cells, resulting in oxidized species of uroporphyrinogen that inhibit the activity of hepatic UROD. It is also felt to increase the uptake of iron in liver cells, leading to further oxidation of uroporphyrinogen by the release of activated oxygen species. Additionally, exposure to chlorinated cyclic hydrocarbons can lead to a deficiency in the activity of uroporphyrinogen decarboxylase, causing the buildup of excess uroporphyrinogen. Additionally, alcohol has been shown to increase the activity of the delta-aminolevulinic acid synthetase (ALA synthetase), the rate-limiting enzymatic step in heme synthesis in the mitochondria, in rats.[24] Therefore, alcohol consumption may increase the production of uroporphyrinogen, exacerbating symptoms in individuals with porphyria cutanea tarda.[citation needed]
Diagnosis
While the most common symptom of PCT is the appearance of skin lesions and blistering, their appearance is not conclusive. Laboratory testing commonly reveals high levels of uroporphyrinogen in the urine, clinically referred to as uroporphyrinogenuria. Additionally, testing for common risk factors such as hepatitis C and hemochromatosis is strongly suggested, as their high prevalence in patients with PCT may require additional treatment. If the clinical appearance of PCT is present, but the laboratories are negative, the diagnosis of pseudoporphyria should be seriously considered.[citation needed]
Classification
Some sources divide PCT into two types: sporadic and familial.[25] Other sources include a third type,[26] but this is less common.
Type I porphyria cutanea tarda, the sporadic form, is indicated by UROD deficiency that is observed only in hepatic cells and nowhere else in the body. Genetically, these individuals will not exhibit deficiency in the UROD gene, although other genetic factors, such as HFE deficiency (resulting in hemochromatosis and the buildup of iron in the liver), are thought to play a key role. Typically, in these individuals, a variety of risk factors such as alcohol use disorder and Hepatitis C virus infection co-occur to result in the clinical manifestation of PCT.
Patients exhibiting Type II PCT have a specific deficiency in the UROD gene, passed down in an autosomal dominant pattern. Those possessing this deficiency are heterozygous for the UROD gene. They do not show a complete lack of functional uroporphyrinogen decarboxylase, only a deficient form of the enzyme that is marked by reduced conversion of uroporphyrinogen to coproporphyrinogen. Therefore, the expression of uroporphyrinogen decarboxylase will be reduced throughout the body of these individuals, while it is isolated to the liver in Type I patients. While this genetic deficiency is the main distinction between Type I and Type II PCT, the risk factors mentioned before are often seen in patients presenting with Type II PCT. Many people who possess the deficient UROD gene often go their entire lives without having a clinical manifestation of PCT symptoms.
Type III
-
The least common is Type III, which is no different from Type I insofar as the patients possess normal UROD genes. Despite this, Type III PCT is observed in more than one family member, indicating a genetic component unrelated to the expression of uroporphyrinogen decarboxylase.
One study used 74% as the cutoff for UROD activity, with those patients under that number being classified as type II, and those above classified as type III if there was a family history, and type I if there was not.[27]
Genetic variants associated with hemochromatosis have been observed in PCT patients,[13] which may help explain inherited PCT not associated with UROD.
Treatment
Since PCT is a chronic condition, comprehensive management of the disease is the most effective form of treatment. Primarily, it is key that patients diagnosed with PCT avoid alcohol consumption, iron supplements, excess exposure to sunlight (especially in the summer), as well as estrogen and chlorinated cyclic hydrocarbons, all of which can potentially exacerbate the disorder. Additionally, the management of excess iron (due to the commonality of hemochromatosis in PCT patients) can be achieved through phlebotomy, whereby blood is systematically drained from the patient. A borderline iron deficiency has been found to have a protective effect by limiting heme synthesis. In the absence of iron, which is to be incorporated in the porphyrin formed in the last step of the synthesis, the mRNA of erythroid 5-aminolevulinate synthase (ALAS-2) is blocked by attachment of an iron-responsive element (IRE) binding cytosolic protein, and transcription of this key enzyme is inhibited.[28]
Low doses of antimalarials can be used.[29] Orally ingested chloroquine is completely absorbed in the gut and is preferentially concentrated in the liver, spleen, and kidneys.[30] They work by removing excess porphyrins from the liver via increasing the excretion rate by forming a coordination complex with the iron center of the porphyrin as well as an intramolecular hydrogen bond between a propionate side chain of the porphyrin and the protonated quinuclidine nitrogen atom of either alkaloid.[31] Due to the presence of the chlorine atom, the entire complex is more water-soluble allowing the kidneys to preferentially remove it from the blood stream and expel it through urination.[30][32][33] Chloroquine treatment can induce porphyria attacks within the first couple of months of treatment due to the mass mobilization of porphyrins from the liver into the bloodstream.[30] Complete remission can be seen within 6–12 months as each dose of antimalarial can only remove a finite amount of porphyrins, and there are generally decades of accumulation to be cleared. Originally, higher doses were used to treat the condition, but are no longer recommended because of liver toxicity.[34][35] Finally, due to the strong association between PCT and Hepatitis C, the treatment of Hepatitis C (if present) is vital to the effective treatment of PCT. Chloroquine, hydroxychloroquine, and venesection are typically employed in the management strategy.[36]
Epidemiology
PCT prevalence is estimated at 1 in 10,000.[37] An estimated 80% of porphyria cutanea tarda cases are sporadic. The exact frequency is not clear because many people with the condition never experience symptoms, and those who do are often misdiagnosed with anything ranging from idiopathicphotodermatitis and seasonal allergies to hives.[citation needed]
↑Di Padova, C.; Marchesi, L.; Cainelli, T.; Gori, G.; Podenzani, S.A.; Rovagnati, P.; Rizzardini, M.; Cantoni, L. (1983). "Effects of Phlebotomy on Urinary Porphyrin Pattern and Liver Histology in Patients with Porphyria Cutanea Tarda". The American Journal of the Medical Sciences. 285 (1): 2–12. doi:10.1097/00000441-198301000-00001. PMID6824014. S2CID23741767.
↑Goljan, E. F. (2011). Pathology (3rd ed., rev. reprint.). Philadelphia, PA: Mosby/Elsevier.[pageneeded]
12Rocchi, E.; Stella, A. M.; Cassanelli, M.; Borghi, A.; Nardella, N.; Seium, Y.; Casalgrandi, G. (1 July 1995). "Liposoluble vitamins and naturally occurring carotenoids in porphyria cutanea tarda". European Journal of Clinical Investigation. 25 (7): 510–514. doi:10.1111/j.1365-2362.1995.tb01737.x. PMID7556369. S2CID44998779.
↑Rocchi, E.; Casalgrandi, G.; Masini, A.; Giovannini, F.; Ceccarelli, D.; Ferrali, M.; Marchini, S.; Ventura, E. (1 December 1999). "Circulating pro- and antioxidant factors in iron and porphyrin metabolism disorders". Italian Journal of Gastroenterology and Hepatology. 31 (9): 861–867. PMID10669994.
12Benoldi, D.; Manfredi, G.; Pezzarossa, E.; Allegra, F. (1 December 1981). "Retinol binding protein in normal human skin and in cutaneous disorders". The British Journal of Dermatology. 105 (6): 659–665. doi:10.1111/j.1365-2133.1981.tb00976.x. PMID7032574. S2CID23170672.
↑Böhm, F.; Edge, R.; Foley, S.; Lange, L.; Truscott, T. G. (31 December 2001). "Antioxidant inhibition of porphyrin-induced cellular phototoxicity". Journal of Photochemistry and Photobiology B: Biology. 65 (2–3): 177–183. Bibcode:2001JPPB...65..177B. doi:10.1016/s1011-1344(01)00259-7. PMID11809377.
↑Jackson, A. H.; Ferramola, A. M.; Sancovich, H. A.; Evans, N; Matlin, S. A.; Ryder, D. J.; Smith, S. G. (1976). "Hepta- and hexa-carboxylic porphyrinogen intermediates in haem biosynthesis". Annals of Clinical Research. 8 (Suppl 17): 64–9. PMID1008499.
↑Miller, Dennis M.; Woods, James S. (1993). "Urinary porphyrins as biological indicators of oxidative stress in the kidney". Biochemical Pharmacology. 46 (12): 2235–41. doi:10.1016/0006-2952(93)90614-3. PMID8274157.
↑Brun, Atle; Sandberg, Sverre (1991). "Mechanisms of photosensitivity in porphyric patients with special emphasis on erythropoietic protoporphyria". Journal of Photochemistry and Photobiology B: Biology. 10 (4): 285–302. Bibcode:1991JPPB...10..285B. doi:10.1016/1011-1344(91)80015-A. PMID1791486.
↑Lim, H. W. (1989). "Mechanisms of phototoxicity in porphyria cutanea tarda and erythropoietic protoporphyria". Immunology Series. 46: 671–85. PMID2488874.
↑Held, H. (2009). "Effect of Alcohol on the Heme and Porphyrin Synthesis Interaction with Phenobarbital and Pyrazole". Digestion. 15 (2): 136–46. doi:10.1159/000197995. PMID838185.
↑Méndez, M.; Poblete-Gutiérrez, P.; García-Bravo, M.; Wiederholt, T.; Morán-Jiménez, M.J.; Merk, H.F.; Garrido-Astray, M.C.; Frank, J.; Fontanellas, A.; Enríquez De Salamanca, R. (2007). "Molecular heterogeneity of familial porphyria cutanea tarda in Spain: Characterization of 10 novel mutations in the UROD gene". British Journal of Dermatology. 157 (3): 501–7. doi:10.1111/j.1365-2133.2007.08064.x. hdl:11268/5436. PMID17627795. S2CID43785629.
↑Cruz-Rojo, J; Fontanellas, A; Morán-Jiménez, M. J.; Navarro-Ordóñez, S; García-Bravo, M; Méndez, M; Muñoz-Rivero, M. C.; De Salamanca, R. E. (2002). "Precipitating/aggravating factors of porphyria cutanea tarda in Spanish patients". Cellular and Molecular Biology (Noisy-le-Grand, France). 48 (8): 845–52. PMID12699242.
↑Thunell, S (2000). "Porphyrins, porphyrin metabolism and porphyrias. I. Update". Scandinavian Journal of Clinical and Laboratory Investigation. 60 (7): 509–40. doi:10.1080/003655100448310. PMID11202048. S2CID8470078.
↑De Villiers, Katherine A.; Gildenhuys, Johandie; Le Roex, Tanya (2012). "Iron(III) Protoporphyrin IX Complexes of the Antimalarial CinchonaAlkaloids Quinine and Quinidine". ACS Chemical Biology. 7 (4): 666–71. doi:10.1021/cb200528z. PMID22276975.
↑Asghari-Khiavi, Mehdi; Vongsvivut, Jitraporn; Perepichka, Inna; Mechler, Adam; Wood, Bayden R.; McNaughton, Don; Bohle, D. Scott (2011). "Interaction of quinoline antimalarial drugs with ferriprotoporphyrin IX, a solid state spectroscopy study". Journal of Inorganic Biochemistry. 105 (12): 1662–9. doi:10.1016/j.jinorgbio.2011.08.005. PMID22079977.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.