Database of protein families, domains and functional sites
InterPro
Content
Description
InterPro functionally analyzes protein sequences and classifies them into protein families while predicting the presence of domains and functional sites.
InterPro is a database of protein families, protein domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences[2] in order to functionally characterise them.[3][4]
The contents of InterPro consist of diagnostic signatures and the proteins that they significantly match. The signatures consist of models (simple types, such as regular expressions or more complex ones, such as Hidden Markov models) which describe protein families, domains or sites. Unknown sequences are searched to create homology models. Each of the member databases of InterPro contributes towards a different niche, from very high-level, structure-based classifications (SUPERFAMILY and CATH-Gene3D) through to quite specific sub-family classifications (PRINTS and PANTHER).
InterPro's intention is to provide a one-stop-shop for protein classification, where all the signatures produced by the different member databases are placed into entries within the InterPro database. Signatures which represent equivalent domains, sites or families are put into the same entry and entries can also be related to one another. Additional information such as a description, consistent names and Gene Ontology (GO) terms are associated with each entry, where possible.
Data contained in InterPro
InterPro contains three main entities: proteins, signatures (also referred to as "methods" or "models") and entries. The proteins in UniProtKB are also the central protein entities in InterPro. Information regarding which signatures significantly match these proteins are calculated as the sequences are released by UniProtKB and these results are made available to the public (see below). The matches of signatures to proteins are what determine how signatures are integrated together into InterPro entries: comparative overlap of matched protein sets and the location of the signatures' matches on the sequences are used as indicators of relatedness. Only signatures deemed to be of sufficient quality are integrated into InterPro. As of version 81.0 (released 21 August 2020) InterPro entries annotated 73.9% of residues found in UniProtKB with another 9.2% annotated by signatures that are pending integration.[5]
The coverage of UniProtKB residues by InterPro entries as of InterPro version 81.0.
InterPro also includes data for splice variants and the proteins contained in the UniParc and UniMES databases.
InterPro consortium member databases
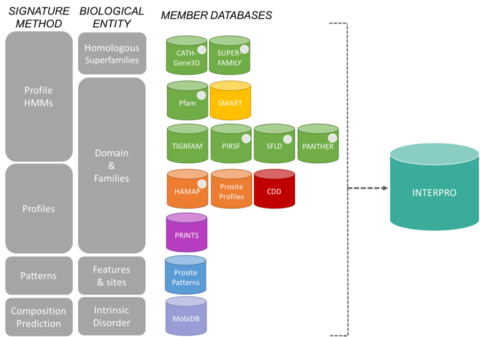
The signatures from InterPro come from 13 "member databases", which are listed below.
CATH-Gene3D
Describes protein families and domain architectures in complete genomes. Protein families are formed using a Markov clustering algorithm, followed by multi-linkage clustering according to sequence identity. Mapping of predicted structure and sequence domains is undertaken using hidden Markov models libraries representing CATH and Pfam domains. Functional annotation is provided to proteins from multiple resources. Functional prediction and analysis of domain architectures is available from the Gene3D website.
CDD
Conserved Domain Database is a protein annotation resource that consists of a collection of annotated multiple sequence alignment models for ancient domains and full-length proteins. These are available as position-specific score matrices (PSSMs) for fast identification of conserved domains in protein sequences via RPS-BLAST.
HAMAP
Stands for High-quality Automated and Manual Annotation of microbial Proteomes. HAMAP profiles are manually created by expert curators they identify proteins that are part of well-conserved bacterial, archaeal and plastid-encoded (i.e. chloroplasts, cyanelles, apicoplasts, non-photosynthetic plastids) proteins families or subfamilies.
MobiDB
MobiDB is database annotating intrinsic disorder in proteins.
PANTHER
PANTHER is a large collection of protein families that have been subdivided into functionally related subfamilies, using human expertise. These subfamilies model the divergence of specific functions within protein families, allowing more accurate association with function (human-curated molecular function and biological process classifications and pathway diagrams), as well as inference of amino acids important for functional specificity. Hidden Markov models (HMMs) are built for each family and subfamily for classifying additional protein sequences.
Pfam
Is large collection of multiple sequence alignments and hidden Markov models covering many common protein domains and families. The 13 member databases of the InterPro consortium grouped by their signature construction method and the biological entity they focus on.
PIRSF
Protein classification system is a network with multiple levels of sequence diversity from superfamilies to subfamilies that reflects the evolutionary relationship of full-length proteins and domains. The primary PIRSF classification unit is the homeomorphic family, whose members are both homologous (evolved from a common ancestor) and homeomorphic (sharing full-length sequence similarity and a common domain architecture).
PRINTS
PRINTS is a compendium of protein fingerprints. A fingerprint is a group of conserved motifs used to characterise a protein family; its diagnostic power is refined by iterative scanning of UniProt. Usually the motifs do not overlap, but are separated along a sequence, though they may be contiguous in 3D-space. Fingerprints can encode protein folds and functionalities more flexibly and powerfully than can single motifs, their full diagnostic potency deriving from the mutual context afforded by motif neighbours.
PROSITE
PROSITE is a database of protein families and domains. It consists of biologically significant sites, patterns and profiles that help to reliably identify to which known protein family (if any) a new sequence belongs.
SMART
Simple Modular Architecture Research Tool Allows the identification and annotation of genetically mobile domains and the analysis of domain architectures. More than 800 domain families found in signaling, extracellular and chromatin-associated proteins are detectable. These domains are extensively annotated with respect to phyletic distributions, functional class, tertiary structures and functionally important residues.
SUPERFAMILY
SUPERFAMILY is a library of profile hidden Markov models that represent all proteins of known structure. The library is based on the SCOP classification of proteins: each model corresponds to a SCOP domain and aims to represent the entire SCOP superfamily that the domain belongs to. SUPERFAMILY has been used to carry out structural assignments to all completely sequenced genomes.
SFLD
A hierarchical classification of enzymes that relates specific sequence-structure features to specific chemical capabilities.
TIGRFAMs
TIGRFAMs is a collection of protein families, featuring curated multiple sequence alignments, hidden Markov models (HMMs) and annotation, which provides a tool for identifying functionally related proteins based on sequence homology. Those entries which are "equivalogs" group homologous proteins which are conserved with respect to function.
Data types
InterPro consists of seven types of data provided by different members of the consortium:
Data Types of InterPro
Data Type
Description
Contributing Databases
InterPro Entries
Structural and/or functional domains of proteins predicted using one or more signatures
All 13 member databases
Member Database signatures
Signatures from member databases. These include signatures that are integrated into InterPro, and those that are not
Icons that identify the five entry types found in InterPro (Homologous Superfamily, Family, Domain, Repeat, or Site).
InterPro entry types
InterPro entries can be further broken down into five types:
Homologous Superfamily: A group of proteins that share a common evolutionary origin as seen in their structural similarities, even if their sequences are not highly similar. These entries are specifically only provided by two member databases: CATH-Gene3D and SUPERFAMILY.
Family: A group of proteins that have a common evolutionary origin determined through structural similarities, related functions, or sequence homology.
Domain: A distinct unit in a protein with a particular function, structure, or sequence.
Repeat: A sequence of amino acids, usually no longer than 50 amino acids, that tend to repeat many times in a protein.
The database is available for text- and sequence-based searches via a webserver, and for download via anonymous FTP. Like other EBI databases, it is in the public domain, since its content can be used "by any individual and for any purpose".[8] InterPro aims to release data to the public every 8 weeks, typically within a day of the UniProtKB release of the same proteins.
InterPro application programming interface (API)
InterPro provides an API for programmatic access to all InterPro entries and their related entries in Json format.[9] There are six main endpoints for the API corresponding to the different InterPro data types: entry, protein, structure, taxonomy, proteome and set.
InterProScan
InterProScan is a software package that allows users to scan sequences against member database signatures. Users can use this signature scanning software to functionally characterize novel nucleotide or protein sequences.[10] InterProScan is frequently used in genome projects in order to obtain a "first-pass" characterisation of the genome of interest.[11][12]As of December2020[update], the public version of InterProScan (v5.x) uses a Java-based architecture.[13] The software package is currently only supported on a 64-bit Linux operating system.
InterProScan, along with many other EMBL-EBI bioinformatics tools, can also be accessed programmatically using RESTful and SOAP Web Services APIs.[14]
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.