Related Research Articles
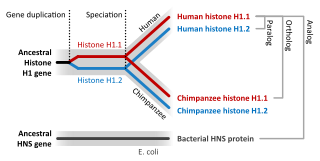
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).

UniProt is a freely accessible database of protein sequence and functional information, many entries being derived from genome sequencing projects. It contains a large amount of information about the biological function of proteins derived from the research literature. It is maintained by the UniProt consortium, which consists of several European bioinformatics organisations and a foundation from Washington, DC, United States.
The Rat Genome Database (RGD) is a database of rat genomics, genetics, physiology and functional data, as well as data for comparative genomics between rat, human and mouse. RGD is responsible for attaching biological information to the rat genome via structured vocabulary, or ontology, annotations assigned to genes and quantitative trait loci (QTL), and for consolidating rat strain data and making it available to the research community. They are also developing a suite of tools for mining and analyzing genomic, physiologic and functional data for the rat, and comparative data for rat, mouse, human, and five other species.
KEGG is a collection of databases dealing with genomes, biological pathways, diseases, drugs, and chemical substances. KEGG is utilized for bioinformatics research and education, including data analysis in genomics, metagenomics, metabolomics and other omics studies, modeling and simulation in systems biology, and translational research in drug development.
InterPro is a database of protein families, protein domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.
The Saccharomyces Genome Database (SGD) is a scientific database of the molecular biology and genetics of the yeast Saccharomyces cerevisiae, which is commonly known as baker's or budding yeast. Further information is located at the Yeastract curated repository.

The Pathogen-Host Interactions database (PHI-base) is a biological database that contains manually curated information on genes experimentally proven to affect the outcome of pathogen-host interactions. The database has been maintained by researchers at Rothamsted Research and external collaborators since 2005. PHI-base has been part of the UK node of ELIXIR, the European life-science infrastructure for biological information, since 2016.
FlyBase is an online bioinformatics database and the primary repository of genetic and molecular data for the insect family Drosophilidae. For the most extensively studied species and model organism, Drosophila melanogaster, a wide range of data are presented in different formats.

MicrobesOnline is a publicly and freely accessible website that hosts multiple comparative genomic tools for comparing microbial species at the genomic, transcriptomic and functional levels. MicrobesOnline was developed by the Virtual Institute for Microbial Stress and Survival, which is based at the Lawrence Berkeley National Laboratory in Berkeley, California. The site was launched in 2005, with regular updates until 2011.
SUPERFAMILY is a database and search platform of structural and functional annotation for all proteins and genomes. It classifies amino acid sequences into known structural domains, especially into SCOP superfamilies. Domains are functional, structural, and evolutionary units that form proteins. Domains of common Ancestry are grouped into superfamilies. The domains and domain superfamilies are defined and described in SCOP. Superfamilies are groups of proteins which have structural evidence to support a common evolutionary ancestor but may not have detectable sequence homology.
Protein function prediction methods are techniques that bioinformatics researchers use to assign biological or biochemical roles to proteins. These proteins are usually ones that are poorly studied or predicted based on genomic sequence data. These predictions are often driven by data-intensive computational procedures. Information may come from nucleic acid sequence homology, gene expression profiles, protein domain structures, text mining of publications, phylogenetic profiles, phenotypic profiles, and protein-protein interaction. Protein function is a broad term: the roles of proteins range from catalysis of biochemical reactions to transport to signal transduction, and a single protein may play a role in multiple processes or cellular pathways.

In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
OrthoDB presents a catalog of orthologous protein-coding genes across vertebrates, arthropods, fungi, plants, and bacteria. Orthology refers to the last common ancestor of the species under consideration, and thus OrthoDB explicitly delineates orthologs at each major radiation along the species phylogeny. The database of orthologs presents available protein descriptors, together with Gene Ontology and InterPro attributes, which serve to provide general descriptive annotations of the orthologous groups, and facilitate comprehensive orthology database querying. OrthoDB also provides computed evolutionary traits of orthologs, such as gene duplicability and loss profiles, divergence rates, sibling groups, and gene intron-exon architectures.
PhylomeDB is a public biological database for complete catalogs of gene phylogenies (phylomes). It allows users to interactively explore the evolutionary history of genes through the visualization of phylogenetic trees and multiple sequence alignments. Moreover, phylomeDB provides genome-wide orthology and paralogy predictions which are based on the analysis of the phylogenetic trees. The automated pipeline used to reconstruct trees aims at providing a high-quality phylogenetic analysis of different genomes, including Maximum Likelihood tree inference, alignment trimming and evolutionary model testing.
The human gene Chromosome 3 open reading frame 14 is a gene of uncertain function located at 3p14.2 near fragile site FRBA3—which falls between this gene and the centromere. Its protein is expected to localize to the nucleus and bind DNA. Orthologs have been identified in all of the major animal groups, minus amphibians and insects, tracing as far back as the sea anemone; indicating an origin of over 1000 mya, highlighting its importance in the animal genome.
DisProt is a manually curated biological database of intrinsically disordered proteins (IDPs) and regions (IDRs). DisProt annotations cover state information on the protein but also, when available, its state transitions, interactions and functional aspects of disorder detected by specific experimental methods. DisProt is hosted and maintained in the BioComputing UP laboratory.
dcGO is a comprehensive ontology database for protein domains. As an ontology resource, dcGO integrates Open Biomedical Ontologies from a variety of contexts, ranging from functional information like Gene Ontology to others on enzymes and pathways, from phenotype information across major model organisms to information about human diseases and drugs. As a protein domain resource, dcGO includes annotations to both the individual domains and supra-domains.
PomBase is a model organism database that provides online access to the fission yeast Schizosaccharomyces pombe genome sequence and annotated features, together with a wide range of manually curated functional gene-specific data. The PomBase website was redeveloped in 2016 to provide users with a more fully integrated, better-performing service.
Model organism databases (MODs) are biological databases, or knowledgebases, dedicated to the provision of in-depth biological data for intensively studied model organisms. MODs allow researchers to easily find background information on large sets of genes, plan experiments efficiently, combine their data with existing knowledge, and construct novel hypotheses. They allow users to analyse results and interpret datasets, and the data they generate are increasingly used to describe less well studied species. Where possible, MODs share common approaches to collect and represent biological information. For example, all MODs use the Gene Ontology (GO) to describe functions, processes and cellular locations of specific gene products. Projects also exist to enable software sharing for curation, visualization and querying between different MODs. Organismal diversity and varying user requirements however mean that MODs are often required to customize capture, display, and provision of data.
References
- 1 2 3 Thomas, PD.; Kejariwal, A.; Campbell, MJ.; Mi, H.; Diemer, K.; Guo, N.; Ladunga, I.; Ulitsky-Lazareva, B.; et al. (Jan 2003). "PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification". Nucleic Acids Res. 31 (1): 334–41. doi:10.1093/nar/gkg115. PMC 165562 . PMID 12520017.
- 1 2 "GO Reference Genome Annotation Project".
- 1 2 3 4 5 6 7 8 Mi, H.; Muruganujan, A.; Thomas, PD. (Jan 2013). "PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees". Nucleic Acids Res. 41 (Database issue): D377–86. doi:10.1093/nar/gks1118. PMC 3531194 . PMID 23193289.
- 1 2 Hunter, S.; Jones, P.; Mitchell, A.; Apweiler, R.; Attwood, TK.; Bateman, A.; Bernard, T.; Binns, D.; et al. (Jan 2012). "InterPro in 2011: new developments in the family and domain prediction database". Nucleic Acids Res. 40 (Database issue): D306–12. doi:10.1093/nar/gkr948. PMC 3245097 . PMID 22096229.
- ↑ Mi, H.; Muruganujan, A.; Thomas, PD. (Aug 2013). "Large-scale gene function analysis with the PANTHER classification system". Nucleic Acids Res. 8 (8): 1551–66. doi:10.1038/nprot.2013.092. PMC 6519453 . PMID 23868073.
- 1 2 3 "PANTHERdb".
- ↑ Mi, H; Huang, X; Muruganujan, A; Tang, H; Mills, C; Kang, D; Thomas, PD (29 November 2016). "PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements". Nucleic Acids Research. 45 (D1): D183–D189. doi:10.1093/nar/gkw1138. PMC 5210595 . PMID 27899595.
- ↑ The UniProt Consortium (Jan 2012). "Reorganizing the protein space at the Universal Protein Resource (UniProt)". Nucleic Acids Res. 40 (D1): D71–D75. doi:10.1093/nar/gkr981. PMC 3245120 . PMID 22102590.
- ↑ Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. (Sep 2011). "Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium". Brief Bioinform. 12 (5): 449–62. doi:10.1093/bib/bbr042. PMC 3178059 . PMID 21873635.
- ↑ Gene Ontology Consortium (Jan 2012). "The Gene Ontology: enhancements for 2011". Nucleic Acids Res. 40 (D1): D559–D564. doi:10.1093/nar/gkr1028. PMC 3245151 . PMID 22102568.
- ↑ Wu, C.H.; Apweiler, R.; Bairoch, A.; Natale, D.A.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; et al. (Jan 2006). "The Universal Protein Resource (UniProt): an expanding universe of protein information". Nucleic Acids Res. 34 (Database issue): D187–D191. doi:10.1093/nar/gkj161. PMC 1347523 . PMID 16381842.
- ↑ Mi, H.; Guo, N.; Thomas, P.D. (Jan 2007). "PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways". Nucleic Acids Res. 35 (Database issue): D247–D252. doi:10.1093/nar/gkl869. PMC 1716723 . PMID 17130144.
- ↑ Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Chen, Y.; Clapham, P.; Coates, G.; et al. (Jan 2011). "Ensembl". Nucleic Acids Res. 39 (Database issue): D800–D806. doi:10.1093/nar/gkq1064. PMC 3013672 . PMID 21045057.
- ↑ Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. (Jan 2011). "Entrez Gene: gene-centered information at NCBI". Nucleic Acids Res. 39 (Database issue): D52–D57. doi:10.1093/nar/gkq1237. PMC 3013746 . PMID 21115458.
- ↑ Mi, H.; Dong, Q.; Muruganujan, A.; Gaudet, P.; Lewis, S.; Thomas, P.D. (Jan 2010). "PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium". Nucleic Acids Res. 38 (Database issue): D204–D210. doi:10.1093/nar/gkp1019. PMC 2808919 . PMID 20015972.
- ↑ "reference proteome".
- ↑ Details in PANTHER 9 statistics can be found here (http://www.pantherdb.org/panther/summaryStats.jsp)