Related Research Articles

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences such as calculating the distance cost between strings in a natural language, or to display financial data.
In bioinformatics and biochemistry, the FASTA format is a text-based format for representing either nucleotide sequences or amino acid (protein) sequences, in which nucleotides or amino acids are represented using single-letter codes.
The printf family of functions in the C programming language are a set of functions that take a format string as input among a variable sized list of other values and produce as output a string that corresponds to the format specifier and given input values. The string is written in a simple template language: characters are usually copied literally into the function's output, but format specifiers, which start with a % character, indicate the location and method to translate a piece of data to characters. The design has been copied to expose similar functionality in other programming languages.
In bioinformatics, sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology might not be able to 'read' whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically, the short fragments (reads) result from shotgun sequencing genomic DNA, or gene transcript (ESTs).
Mach-O, short for Mach object file format, is a file format for executables, object code, shared libraries, dynamically loaded code, and core dumps. It was developed to replace the a.out format.

UCSF Chimera is an extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles. High-quality images and movies can be created. Chimera includes complete documentation and can be downloaded free of charge for noncommercial use.

A Phred quality score is a measure of the quality of the identification of the nucleobases generated by automated DNA sequencing. It was originally developed for the computer program Phred to help in the automation of DNA sequencing in the Human Genome Project. Phred quality scores are assigned to each nucleotide base call in automated sequencer traces. The FASTQ format encodes phred scores as ASCII characters alongside the read sequences. Phred quality scores have become widely accepted to characterize the quality of DNA sequences, and can be used to compare the efficacy of different sequencing methods. Perhaps the most important use of Phred quality scores is the automatic determination of accurate, quality-based consensus sequences.
The Binary Universal Form for the Representation of meteorological data (BUFR) is a binary data format maintained by the World Meteorological Organization (WMO). The latest version is BUFR Edition 4. BUFR Edition 3 is also considered current for operational use. BUFR was created in 1988 with the goal of replacing the WMO's dozens of character-based, position-driven meteorological codes, such as SYNOP, TEMP and CLIMAT. BUFR was designed to be portable, compact, and universal. Any kind of data can be represented, along with its specific spatial/temporal context and any other associated metadata. In the WMO terminology, BUFR belongs to the category of table-driven code forms, where the meaning of data elements is determined by referring to a set of tables that are kept and maintained separately from the message itself.
UGENE is computer software for bioinformatics. It works on personal computer operating systems such as Windows, macOS, or Linux. It is released as free and open-source software, under a GNU General Public License (GPL) version 2.
FASTQ format is a text-based format for storing both a biological sequence and its corresponding quality scores. Both the sequence letter and quality score are each encoded with a single ASCII character for brevity.
The Variant Call Format (VCF) is a standard text file format used in bioinformatics for storing gene sequence variations. The format was developed in 2010 for the 1000 Genomes Project and has since been used by other large-scale genotyping and DNA sequencing projects. VCF is a common output format for variant calling programs due to its relative simplicity and scalability. Many tools have been developed for editing and manipulating VCF files, including VCFtools, which was released in conjunction with the VCF format in 2011, and BCFtools, which was included as part of SAMtools until being split into an independent package in 2014.
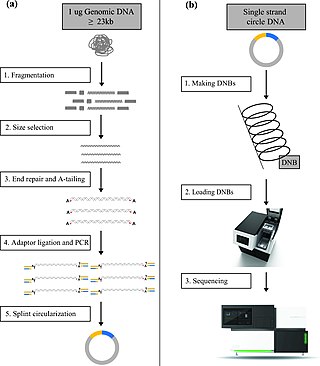
DNA nanoball sequencing is a high throughput sequencing technology that is used to determine the entire genomic sequence of an organism. The method uses rolling circle replication to amplify small fragments of genomic DNA into DNA nanoballs. Fluorescent nucleotides bind to complementary nucleotides and are then polymerized to anchor sequences bound to known sequences on the DNA template. The base order is determined via the fluorescence of the bound nucleotides This DNA sequencing method allows large numbers of DNA nanoballs to be sequenced per run at lower reagent costs compared to other next generation sequencing platforms. However, a limitation of this method is that it generates only short sequences of DNA, which presents challenges to mapping its reads to a reference genome. After purchasing Complete Genomics, the Beijing Genomics Institute (BGI) refined DNA nanoball sequencing to sequence nucleotide samples on their own platform.

XQuery API for Java (XQJ) refers to the common Java API for the W3C XQuery 1.0 specification.
Pileup format is a text-based format for summarizing the base calls of aligned reads to a reference sequence. This format facilitates visual display of SNP/indel calling and alignment. It was first used by Tony Cox and Zemin Ning at the Wellcome Trust Sanger Institute, and became widely known through its implementation within the SAMtools software suite.
SAMtools is a set of utilities for interacting with and post-processing short DNA sequence read alignments in the SAM, BAM and CRAM formats, written by Heng Li. These files are generated as output by short read aligners like BWA. Both simple and advanced tools are provided, supporting complex tasks like variant calling and alignment viewing as well as sorting, indexing, data extraction and format conversion. SAM files can be very large, so compression is used to save space. SAM files are human-readable text files, and BAM files are simply their binary equivalent, whilst CRAM files are a restructured column-oriented binary container format. BAM files are typically compressed and more efficient for software to work with than SAM. SAMtools makes it possible to work directly with a compressed BAM file, without having to uncompress the whole file. Additionally, since the format for a SAM/BAM file is somewhat complex - containing reads, references, alignments, quality information, and user-specified annotations - SAMtools reduces the effort needed to use SAM/BAM files by hiding low-level details.
Heng Li is a Chinese bioinformatics scientist. He is an associate professor at the department of Biomedical Informatics of Harvard Medical School and the department of Data Science of Dana-Farber Cancer Institute. He was previously a research scientist working at the Broad Institute in Cambridge, Massachusetts with David Reich and David Altshuler. Li's work has made several important contributions in the field of next generation sequencing.

Binary Alignment Map (BAM) is the comprehensive raw data of genome sequencing; it consists of the lossless, compressed binary representation of the Sequence Alignment Map-files.
Compressed Reference-oriented Alignment Map (CRAM) is a compressed columnar file format for storing biological sequences aligned to a reference sequence, initially devised by Markus Hsi-Yang Fritz et al.
The BED format is a text file format used to store genomic regions as coordinates and associated annotations. The data are presented in the form of columns separated by spaces or tabs. This format was developed during the Human Genome Project and then adopted by other sequencing projects. As a result of this increasingly wide use, this format had already become a de facto standard in bioinformatics before a formal specification was written.
References
- 1 2 3 4 5 Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. (2009). "The Sequence Alignment/Map format and SAMtools" (PDF). Bioinformatics. 25 (16): 2078–2079. doi:10.1093/bioinformatics/btp352. ISSN 1367-4803. PMC 2723002 . PMID 19505943.
- ↑ Edmunds, Scott (2021-02-17). "Play it again, SAMtools. Q&A with the SAMtools team on 12 years of providing bioinformatics "glue"". GigaScience. Retrieved 2021-03-20.
- ↑ Dörpinghaus, J.; Weil, V.; Schaaf, S.; Apke, A. (2023). Computational Life Sciences: Data Engineering and Data Mining for Life Sciences. Studies in Big Data. Springer International Publishing. p. 447. ISBN 978-3-031-08411-9 . Retrieved 2023-07-19.
- 1 2 3 "SAM/BAM Format Specification" (PDF). samtools.github.io.
- ↑ "Explain SAM Flags". broadinstitute.github.io. Retrieved 2023-11-04.