Related Research Articles

In genetics, complementary DNA (cDNA) is DNA that was reverse transcribed from an RNA. cDNA exists in both single-stranded and double-stranded forms and in both natural and engineered forms.

The polymerase chain reaction (PCR) is a method widely used to make millions to billions of copies of a specific DNA sample rapidly, allowing scientists to amplify a very small sample of DNA sufficiently to enable detailed study. PCR was invented in 1983 by American biochemist Kary Mullis at Cetus Corporation. Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.

Reverse transcription polymerase chain reaction (RT-PCR) is a laboratory technique combining reverse transcription of RNA into DNA and amplification of specific DNA targets using polymerase chain reaction (PCR). It is primarily used to measure the amount of a specific RNA. This is achieved by monitoring the amplification reaction using fluorescence, a technique called real-time PCR or quantitative PCR (qPCR). Confusion can arise because some authors use the acronym RT-PCR to denote real-time PCR. In this article, RT-PCR will denote Reverse Transcription PCR. Combined RT-PCR and qPCR are routinely used for analysis of gene expression and quantification of viral RNA in research and clinical settings.
Rapid amplification of cDNA ends (RACE) is a technique used in molecular biology to obtain the full length sequence of an RNA transcript found within a cell. RACE results in the production of a cDNA copy of the RNA sequence of interest, produced through reverse transcription, followed by PCR amplification of the cDNA copies. The amplified cDNA copies are then sequenced and, if long enough, should map to a unique genomic region. RACE is commonly followed up by cloning before sequencing of what was originally individual RNA molecules. A more high-throughput alternative which is useful for identification of novel transcript structures, is to sequence the RACE-products by next generation sequencing technologies.
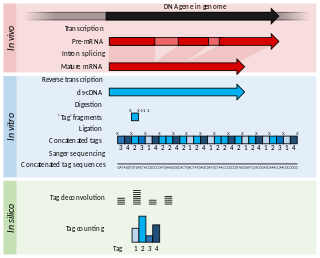
Serial Analysis of Gene Expression (SAGE) is a transcriptomic technique used by molecular biologists to produce a snapshot of the messenger RNA population in a sample of interest in the form of small tags that correspond to fragments of those transcripts. Several variants have been developed since, most notably a more robust version, LongSAGE, RL-SAGE and the most recent SuperSAGE. Many of these have improved the technique with the capture of longer tags, enabling more confident identification of a source gene.
Cross-linking and immunoprecipitation is a method used in molecular biology that combines UV crosslinking with immunoprecipitation in order to identify RNA binding sites of proteins on a transcriptome-wide scale, thereby increasing our understanding of post-transcriptional regulatory networks. CLIP can be used either with antibodies against endogenous proteins, or with common peptide tags or affinity purification, which enables the possibility of profiling model organisms or RBPs otherwise lacking suitable antibodies.
Epigenomics is the study of the complete set of epigenetic modifications on the genetic material of a cell, known as the epigenome. The field is analogous to genomics and proteomics, which are the study of the genome and proteome of a cell. Epigenetic modifications are reversible modifications on a cell's DNA or histones that affect gene expression without altering the DNA sequence. Epigenomic maintenance is a continuous process and plays an important role in stability of eukaryotic genomes by taking part in crucial biological mechanisms like DNA repair. Plant flavones are said to be inhibiting epigenomic marks that cause cancers. Two of the most characterized epigenetic modifications are DNA methylation and histone modification. Epigenetic modifications play an important role in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation/development and tumorigenesis. The study of epigenetics on a global level has been made possible only recently through the adaptation of genomic high-throughput assays.

RNA-Seq is a technique that uses next-generation sequencing to reveal the presence and quantity of RNA molecules in a biological sample, providing a snapshot of gene expression in the sample, also known as transcriptome.
Cap analysis of gene expression (CAGE) is a gene expression technique used in molecular biology to produce a snapshot of the 5′ end of the messenger RNA population in a biological sample. The small fragments from the very beginnings of mRNAs are extracted, reverse-transcribed to cDNA, PCR amplified and sequenced. CAGE was first published by Hayashizaki, Carninci and co-workers in 2003. CAGE has been extensively used within the FANTOM research projects.
iCLIP is a variant of the original CLIP method used for identifying protein-RNA interactions, which uses UV light to covalently bind proteins and RNA molecules to identify RNA binding sites of proteins. This crosslinking step has generally less background than standard RNA immunoprecipitation (RIP) protocols, because the covalent bond formed by UV light allows RNA to be fragmented, followed by stringent purification, and this also enables CLIP to identify the positions of protein-RNA interactions. As with all CLIP methods, iCLIP allows for a very stringent purification of the linked protein-RNA complexes by stringent washing during immunoprecipitation followed by SDS-PAGE and transfer to nitrocellulose. The labelled protein-RNA complexes are then visualised for quality control, excised from nitrocellulose, and treated with proteinase to release the RNA, leaving only a few amino acids at the crosslink site of the RNA.

In the field of cellular biology, single-cell analysis and subcellular analysis is the study of genomics, transcriptomics, proteomics, metabolomics and cell–cell interactions at the single cell level. The concept of single-cell analysis originated in the 1970s. Before the discovery of heterogeneity, single-cell analysis mainly referred to the analysis or manipulation of an individual cell in a bulk population of cells at a particular condition using optical or electronic microscope. To date, due to the heterogeneity seen in both eukaryotic and prokaryotic cell populations, analyzing a single cell makes it possible to discover mechanisms not seen when studying a bulk population of cells. Technologies such as fluorescence-activated cell sorting (FACS) allow the precise isolation of selected single cells from complex samples, while high throughput single cell partitioning technologies, enable the simultaneous molecular analysis of hundreds or thousands of single unsorted cells; this is particularly useful for the analysis of transcriptome variation in genotypically identical cells, allowing the definition of otherwise undetectable cell subtypes. The development of new technologies is increasing our ability to analyze the genome and transcriptome of single cells, as well as to quantify their proteome and metabolome. Mass spectrometry techniques have become important analytical tools for proteomic and metabolomic analysis of single cells. Recent advances have enabled quantifying thousands of protein across hundreds of single cells, and thus make possible new types of analysis. In situ sequencing and fluorescence in situ hybridization (FISH) do not require that cells be isolated and are increasingly being used for analysis of tissues.
Single-cell sequencing examines the nucleic acid sequence information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment. For example, in cancer, sequencing the DNA of individual cells can give information about mutations carried by small populations of cells. In development, sequencing the RNAs expressed by individual cells can give insight into the existence and behavior of different cell types. In microbial systems, a population of the same species can appear genetically clonal. Still, single-cell sequencing of RNA or epigenetic modifications can reveal cell-to-cell variability that may help populations rapidly adapt to survive in changing environments.

A transcriptome in vivo analysis tag is a multifunctional, photoactivatable mRNA-capture molecule designed for isolating mRNA from a single cell in complex tissues.
G&T-seq is a novel form of single cell sequencing technique allowing one to simultaneously obtain both transcriptomic and genomic data from single cells, allowing for direct comparison of gene expression data to its corresponding genomic data in the same cell...

In epitranscriptomic sequencing, most methods focus on either (1) enrichment and purification of the modified RNA molecules before running on the RNA sequencer, or (2) improving or modifying bioinformatics analysis pipelines to call the modification peaks. Most methods have been adapted and optimized for mRNA molecules, except for modified bisulfite sequencing for profiling 5-methylcytidine which was optimized for tRNAs and rRNAs.
Single-cell transcriptomics examines the gene expression level of individual cells in a given population by simultaneously measuring the RNA concentration of hundreds to thousands of genes. Single-cell transcriptomics makes it possible to unravel heterogeneous cell populations, reconstruct cellular developmental pathways, and model transcriptional dynamics — all previously masked in bulk RNA sequencing.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.

Spatial transcriptomics is a method for assigning cell types to their locations in the histological sections and can also be used to determine subcellular localization of mRNA molecules. First described in 2016 by Ståhl et al., it has since undergone a variety of improvements and modifications.
CITE-Seq is a method for performing RNA sequencing along with gaining quantitative and qualitative information on surface proteins with available antibodies on a single cell level. So far, the method has been demonstrated to work with only a few proteins per cell. As such, it provides an additional layer of information for the same cell by combining both proteomics and transcriptomics data. For phenotyping, this method has been shown to be as accurate as flow cytometry by the groups that developed it. It is currently one of the main methods, along with REAP-Seq, to evaluate both gene expression and protein levels simultaneously in different species.
BLESS, also known as breaks labeling, enrichment on streptavidin and next-generation sequencing, is a method used to detect genome-wide double-strand DNA damage. In contrast to chromatin immunoprecipitation (ChIP)-based methods of identifying DNA double-strand breaks (DSBs) by labeling DNA repair proteins, BLESS utilizes biotinylated DNA linkers to directly label genomic DNA in situ which allows for high-specificity enrichment of samples on streptavidin beads and the subsequent sequencing-based DSB mapping to nucleotide resolution.
References
- ↑ König, Julian; Zarnack, Kathi; Rot, Gregor; Curk, Tomaz; Kayikci, Melis; Zupan, Blaz; Turner, Daniel J.; Luscombe, Nicholas M.; Ule, Jernej (July 2010). "iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution". Nature Structural & Molecular Biology. 17 (7): 909–915. doi:10.1038/nsmb.1838. ISSN 1545-9985. PMC 3000544 . PMID 20601959.
- ↑ Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J (2012). "Counting absolute numbers of molecules using unique molecular identifiers". Nat. Methods. 9 (1): 72–4. doi:10.1038/nmeth.1778. PMID 22101854. S2CID 39225091.
- ↑ Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lönnerberg P, Linnarsson S (2014). "Quantitative single-cell RNA-seq with unique molecular identifiers". Nat. Methods. 11 (2): 163–6. doi:10.1038/nmeth.2772. PMID 24363023. S2CID 6765530.
| | This genetics article is a stub. You can help Wikipedia by expanding it. |