Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences such as calculating the distance cost between strings in a natural language, or to display financial data.

The National Center for Biotechnology Information (NCBI) is part of the United States National Library of Medicine (NLM), a branch of the National Institutes of Health (NIH). It is approved and funded by the government of the United States. The NCBI is located in Bethesda, Maryland, and was founded in 1988 through legislation sponsored by US Congressman Claude Pepper.
In bioinformatics, sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of a wide range of analytical methods to understand its features, function, structure, or evolution. Methodologies used include sequence alignment, searches against biological databases, and others.
In bioinformatics, BLAST is an algorithm and program for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA and/or RNA sequences. A BLAST search enables a researcher to compare a subject protein or nucleotide sequence with a library or database of sequences, and identify database sequences that resemble the query sequence above a certain threshold. For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene; BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence.
In bioinformatics, sequence clustering algorithms attempt to group biological sequences that are somehow related. The sequences can be either of genomic, "transcriptomic" (ESTs) or protein origin. For proteins, homologous sequences are typically grouped into families. For EST data, clustering is important to group sequences originating from the same gene before the ESTs are assembled to reconstruct the original mRNA.
FASTA is a DNA and protein sequence alignment software package first described by David J. Lipman and William R. Pearson in 1985. Its legacy is the FASTA format which is now ubiquitous in bioinformatics.
In molecular biology, open reading frames (ORFs) are defined as spans of DNA sequence between the start and stop codons. Usually, this is considered within a studied region of a prokaryotic DNA sequence, where only one of the six possible reading frames will be "open". Such an ORF may contain a start codon and by definition cannot extend beyond a stop codon. That start codon indicates where translation may start. The transcription termination site is located after the ORF, beyond the translation stop codon. If transcription were to cease before the stop codon, an incomplete protein would be made during translation.
A sequence profiling tool in bioinformatics is a type of software that presents information related to a genetic sequence, gene name, or keyword input. Such tools generally take a query such as a DNA, RNA, or protein sequence or ‘keyword’ and search one or more databases for information related to that sequence. Summaries and aggregate results are provided in standardized format describing the information that would otherwise have required visits to many smaller sites or direct literature searches to compile. Many sequence profiling tools are software portals or gateways that simplify the process of finding information about a query in the large and growing number of bioinformatics databases. The access to these kinds of tools is either web based or locally downloadable executables.
The European Bioinformatics Institute (EMBL-EBI) is an intergovernmental organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.
Computational genomics refers to the use of computational and statistical analysis to decipher biology from genome sequences and related data, including both DNA and RNA sequence as well as other "post-genomic" data. These, in combination with computational and statistical approaches to understanding the function of the genes and statistical association analysis, this field is also often referred to as Computational and Statistical Genetics/genomics. As such, computational genomics may be regarded as a subset of bioinformatics and computational biology, but with a focus on using whole genomes to understand the principles of how the DNA of a species controls its biology at the molecular level and beyond. With the current abundance of massive biological datasets, computational studies have become one of the most important means to biological discovery.
The completion of the human genome sequencing in the early 2000s was a turning point in genomics research. Scientists have conducted series of research into the activities of genes and the genome as a whole. The human genome contains around 3 billion base pairs nucleotide, and the huge quantity of data created necessitates the development of an accessible tool to explore and interpret this information in order to investigate the genetic basis of disease, evolution, and biological processes. The field of genomics has continued to grow, with new sequencing technologies and computational tool making it easier to study the genome.

DGLUCY is a protein that in humans is encoded by the DGLUCY gene.
The Viral Bioinformatics Resource Center (VBRC) is an online resource providing access to a database of curated viral genomes and a variety of tools for bioinformatic genome analysis. This resource was one of eight BRCs funded by NIAID with the goal of promoting research against emerging and re-emerging pathogens, particularly those seen as potential bioterrorism threats. The VBRC is now supported by Dr. Chris Upton at the University of Victoria.

HMMER is a free and commonly used software package for sequence analysis written by Sean Eddy. Its general usage is to identify homologous protein or nucleotide sequences, and to perform sequence alignments. It detects homology by comparing a profile-HMM to either a single sequence or a database of sequences. Sequences that score significantly better to the profile-HMM compared to a null model are considered to be homologous to the sequences that were used to construct the profile-HMM. Profile-HMMs are constructed from a multiple sequence alignment in the HMMER package using the hmmbuild program. The profile-HMM implementation used in the HMMER software was based on the work of Krogh and colleagues. HMMER is a console utility ported to every major operating system, including different versions of Linux, Windows, and macOS.
The UCSC Genome Browser is an online and downloadable genome browser hosted by the University of California, Santa Cruz (UCSC). It is an interactive website offering access to genome sequence data from a variety of vertebrate and invertebrate species and major model organisms, integrated with a large collection of aligned annotations. The Browser is a graphical viewer optimized to support fast interactive performance and is an open-source, web-based tool suite built on top of a MySQL database for rapid visualization, examination, and querying of the data at many levels. The Genome Browser Database, browsing tools, downloadable data files, and documentation can all be found on the UCSC Genome Bioinformatics website.
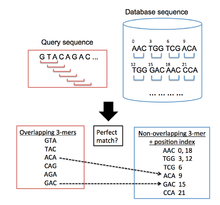
PatternHunter is a commercially available homology search instrument software that uses sequence alignment techniques. It was initially developed in the year 2002 by three scientists: Bin Ma, John Tramp and Ming Li. These scientists were driven by the desire to solve the problem that many investigators face during studies that involve genomics and proteomics. These scientists realized that such studies greatly relied on homology studies that established short seed matches that were subsequently lengthened. Describing homologous genes was an essential part of most evolutionary studies and was crucial to the understanding of the evolution of gene families, the relationship between domains and families. Homologous genes could only be studied effectively using search tools that established like portions or local placement between two proteins or nucleic acid sequences. Homology was quantified by scores obtained from matching sequences, “mismatch and gap scores”.
Non-coding RNAs have been discovered using both experimental and bioinformatic approaches. Bioinformatic approaches can be divided into three main categories. The first involves homology search, although these techniques are by definition unable to find new classes of ncRNAs. The second category includes algorithms designed to discover specific types of ncRNAs that have similar properties. Finally, some discovery methods are based on very general properties of RNA, and are thus able to discover entirely new kinds of ncRNAs.
Bloom filters are space-efficient probabilistic data structures used to test whether an element is a part of a set. Bloom filters require much less space than other data structures for representing sets, however the downside of Bloom filters is that there is a false positive rate when querying the data structure. Since multiple elements may have the same hash values for a number of hash functions, then there is a probability that querying for a non-existent element may return a positive if another element with the same hash values has been added to the Bloom filter. Assuming that the hash function has equal probability of selecting any index of the Bloom filter, the false positive rate of querying a Bloom filter is a function of the number of bits, number of hash functions and number of elements of the Bloom filter. This allows the user to manage the risk of a getting a false positive by compromising on the space benefits of the Bloom filter.