Alpha-ketoglutarate dehydrogenase also known as 2-oxoglutarate dehydrogenase E1 component, mitochondrial is an enzyme that in humans is encoded by the OGDH gene. [5] [6] [7]
Alpha-ketoglutarate dehydrogenase also known as 2-oxoglutarate dehydrogenase E1 component, mitochondrial is an enzyme that in humans is encoded by the OGDH gene. [5] [6] [7]
The OGDH gene is located on the 7th chromosome, with the specific location being 7p14-p13. There are 26 exons located within the gene. [7]
This gene encodes a subunit that catalyzes the oxidative decarboxylation of alpha-ketoglutarate to Succinyl-CoA at its active site in the fourth step of the citric acid cycle by acting as a base to facilitate the decarboxylation. The main residues responsible for the catalysis are thought to be His 260, Phe 227, Gln685, His 729, Ser302, and His 298. [8]
This gene encodes one subunit of the 2-oxoglutarate dehydrogenase complex. This complex catalyzes the overall conversion of 2-oxoglutarate (alpha-ketoglutarate) to succinyl-CoA and CO2 during the citric acid cycle. The protein is located in the mitochondrial matrix and uses thiamine pyrophosphate as a cofactor. [7] The overall complex furthers catalysis by keeping the necessary substrates for the reaction close within the enzyme, thus creating a situation in which it is more likely that the substrate will be in the favorable conformation and orientation. This enzyme is also part of a larger multienzyme complex that channels the intermediates in the catalysis between subunits of the complex thus minimizing unwanted side reactions. Not only do the subunits ferry products back and forth, but each of the subunits in the E1o homodimer are connected via a cavity lined with acidic residues, thus increasing the dimer's ability to act as a base. The orientation of the cavity allows for direct transfer of the intermediate to the E2o subunit. [9]
The protein encoded by OGDH is thought to have a single active site. The enzyme also requires two cofactors in order for it to function properly, Thiamine diphosphate and a divalent magnesium ion. The specific mechanism of the subunit is currently unknown; however, there are several theories as to how it functions, among them is the Hexa Uni Ping Pong theory. [10] Even though the mechanism isn't fully known the kinetic data have been calculated and are as follows: the Km is 0.14 ± 0.04 mM, and the Vmax is 9 ± 3 μmol/(min*mg). [11]
This subunit, known as E1o, catalyzes a rate-limiting step in the citric acid cycle and lies far from equilibrium; the total change in Gibbs free energy is ΔG = −33 kJ/mol. The significant energy change makes it a crucial point of regulation not only for the citric acid cycle, but also for the entire cellular respiration pathway. As such, E1o is inhibited by both NADH and Succinyl-CoA via non competitive feedback inhibition. [8]
A congenital deficiency in 2-oxoglutarate dehydrogenase activity is believed to lead to hypotonia, metabolic acidosis, and hyperlactatemia. It is characterized by the buildup of a chemical called lactic acid in the body and a variety of neurological problems. Signs and symptoms of this condition usually first appear shortly after birth, and they can vary widely among affected individuals. The most common feature is a potentially life-threatening buildup of lactic acid (lactic acidosis), which can cause nausea, vomiting, severe breathing problems, and an abnormal heartbeat. People with pyruvate dehydrogenase deficiency usually have neurological problems as well. Most have delayed development of mental abilities and motor skills such as sitting and walking. Other neurological problems can include intellectual disability, seizures, weak muscle tone (hypotonia), poor coordination, and difficulty walking. Some affected individuals have abnormal brain structures, such as underdevelopment of the tissue connecting the left and right halves of the brain (corpus callosum), wasting away (atrophy) of the exterior part of the brain known as the cerebral cortex, or patches of damaged tissue (lesions) on some parts of the brain. Because of the severe health effects, many individuals with pyruvate dehydrogenase deficiency do not survive past childhood, although some may live into adolescence or adulthood. [7]
Click on genes, proteins and metabolites below to link to respective articles. [§ 1]

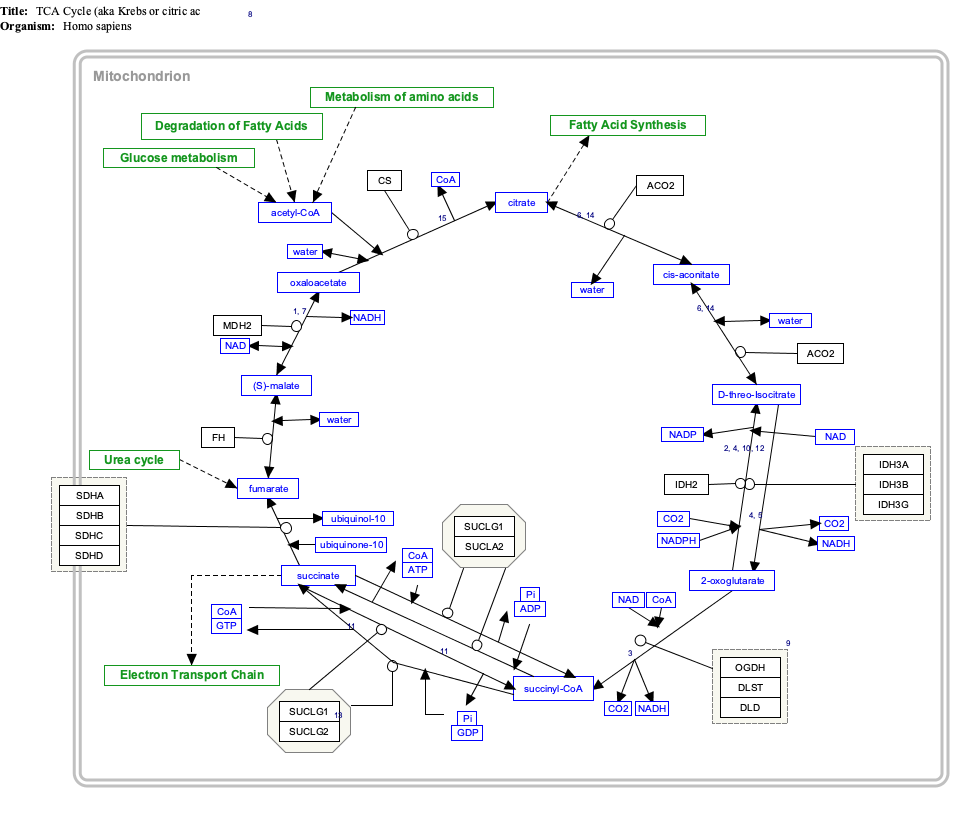
The citric acid cycle (CAC)—also known as the Krebs cycle or the TCA cycle —is a series of chemical reactions to release stored energy through the oxidation of acetyl-CoA derived from carbohydrates, fats, and proteins. The Krebs cycle is used by organisms that respire to generate energy, either by anaerobic respiration or aerobic respiration. In addition, the cycle provides precursors of certain amino acids, as well as the reducing agent NADH, that are used in numerous other reactions. Its central importance to many biochemical pathways suggests that it was one of the earliest components of metabolism and may have originated abiogenically. Even though it is branded as a 'cycle', it is not necessary for metabolites to follow only one specific route; at least three alternative segments of the citric acid cycle have been recognized.

Pyruvate dehydrogenase complex (PDC) is a complex of three enzymes that converts pyruvate into acetyl-CoA by a process called pyruvate decarboxylation. Acetyl-CoA may then be used in the citric acid cycle to carry out cellular respiration, and this complex links the glycolysis metabolic pathway to the citric acid cycle. Pyruvate decarboxylation is also known as the "pyruvate dehydrogenase reaction" because it also involves the oxidation of pyruvate.

Isocitrate dehydrogenase (IDH) (EC 1.1.1.42) and (EC 1.1.1.41) is an enzyme that catalyzes the oxidative decarboxylation of isocitrate, producing alpha-ketoglutarate (α-ketoglutarate) and CO2. This is a two-step process, which involves oxidation of isocitrate (a secondary alcohol) to oxalosuccinate (a ketone), followed by the decarboxylation of the carboxyl group beta to the ketone, forming alpha-ketoglutarate. In humans, IDH exists in three isoforms: IDH3 catalyzes the third step of the citric acid cycle while converting NAD+ to NADH in the mitochondria. The isoforms IDH1 and IDH2 catalyze the same reaction outside the context of the citric acid cycle and use NADP+ as a cofactor instead of NAD+. They localize to the cytosol as well as the mitochondrion and peroxisome.
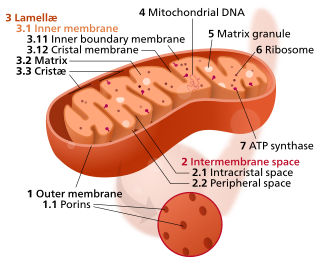
In the mitochondrion, the matrix is the space within the inner membrane. The word "matrix" stems from the fact that this space is viscous, compared to the relatively aqueous cytoplasm. The mitochondrial matrix contains the mitochondrial DNA, ribosomes, soluble enzymes, small organic molecules, nucleotide cofactors, and inorganic ions.[1] The enzymes in the matrix facilitate reactions responsible for the production of ATP, such as the citric acid cycle, oxidative phosphorylation, oxidation of pyruvate, and the beta oxidation of fatty acids.
The oxoglutarate dehydrogenase complex (OGDC) or α-ketoglutarate dehydrogenase complex is an enzyme complex, most commonly known for its role in the citric acid cycle.

Succinyl coenzyme A synthetase is an enzyme that catalyzes the reversible reaction of succinyl-CoA to succinate. The enzyme facilitates the coupling of this reaction to the formation of a nucleoside triphosphate molecule from an inorganic phosphate molecule and a nucleoside diphosphate molecule. It plays a key role as one of the catalysts involved in the citric acid cycle, a central pathway in cellular metabolism, and it is located within the mitochondrial matrix of a cell.

Succinate dehydrogenase [ubiquinone] iron-sulfur subunit, mitochondrial (SDHB) also known as iron-sulfur subunit of complex II (Ip) is a protein that in humans is encoded by the SDHB gene.

Dihydrolipoyl transacetylase is an enzyme component of the multienzyme pyruvate dehydrogenase complex. The pyruvate dehydrogenase complex is responsible for the pyruvate decarboxylation step that links glycolysis to the citric acid cycle. This involves the transformation of pyruvate from glycolysis into acetyl-CoA which is then used in the citric acid cycle to carry out cellular respiration.
Pyruvate dehydrogenase is an enzyme that catalyzes the reaction of pyruvate and a lipoamide to give the acetylated dihydrolipoamide and carbon dioxide. The conversion requires the coenzyme thiamine pyrophosphate.

Dihydrolipoamide dehydrogenase (DLD), also known as dihydrolipoyl dehydrogenase, mitochondrial, is an enzyme that in humans is encoded by the DLD gene. DLD is a flavoprotein enzyme that oxidizes dihydrolipoamide to lipoamide.

Pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial is an enzyme that in humans is encoded by the PDHA1 gene.The pyruvate dehydrogenase complex is a nuclear-encoded mitochondrial matrix multienzyme complex that provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle by catalyzing the irreversible conversion of pyruvate into acetyl-CoA. The PDH complex is composed of multiple copies of 3 enzymes: E1 (PDHA1); dihydrolipoyl transacetylase (DLAT) ; and dihydrolipoyl dehydrogenase (DLD). The E1 enzyme is a heterotetramer of 2 alpha and 2 beta subunits. The E1-alpha subunit contains the E1 active site and plays a key role in the function of the PDH complex.

A 2-oxoisovalerate dehydrogenase subunit alpha, mitochondrial is an enzyme that in humans is encoded by the BCKDHA gene.

Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex, mitochondrial is an enzyme that in humans is encoded by the DLST gene.

2-Oxoisovalerate dehydrogenase subunit beta, mitochondrial is an enzyme that in humans is encoded by the BCKDHB gene.

Isocitrate dehydrogenase [NAD] subunit gamma, mitochondrial is an enzyme that in humans is encoded by the IDH3G gene.

Isocitrate dehydrogenase [NAD] subunit beta, mitochondrial is an enzyme that in humans is encoded by the IDH3B gene.

Succinyl-CoA ligase [GDP-forming] subunit alpha, mitochondrial is an enzyme that in humans is encoded by the SUCLG1 gene.

Malate dehydrogenase, mitochondrial also known as malate dehydrogenase 2 is an enzyme that in humans is encoded by the MDH2 gene.

Pyruvate dehydrogenase (lipoamide) alpha 2, also known as pyruvate dehydrogenase E1 component subunit alpha, testis-specific form, mitochondrial or PDHE1-A type II, is an enzyme that in humans is encoded by the PDHA2 gene.

Pyruvate dehydrogenase (lipoamide) beta, also known as pyruvate dehydrogenase E1 component subunit beta, mitochondrial or PDHE1-B is an enzyme that in humans is encoded by the PDHB gene. The pyruvate dehydrogenase (PDH) complex is a nuclear-encoded mitochondrial multienzyme complex that catalyzes the overall conversion of pyruvate to acetyl-CoA and CO2, and provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle. The PDH complex is composed of multiple copies of three enzymatic components: pyruvate dehydrogenase (E1), dihydrolipoamide acetyltransferase (E2) and lipoamide dehydrogenase (E3). The E1 enzyme is a heterotetramer of two alpha and two beta subunits. This gene encodes the E1 beta subunit. Mutations in this gene are associated with pyruvate dehydrogenase E1-beta deficiency.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.
PDB gallery | |
|---|---|
|