
Medium-chain acyl-CoA dehydrogenase deficiency is a disorder of fatty acid oxidation that impairs the body's ability to break down medium-chain fatty acids into acetyl-CoA. The disorder is characterized by hypoglycemia and sudden death without timely intervention, most often brought on by periods of fasting or vomiting.

Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency is a rare autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats into energy. This can become life-threatening, particularly during periods of fasting.
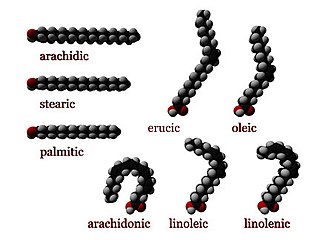
Numerous genetic disorders are caused by errors in fatty acid metabolism. These disorders may be described as fatty oxidation disorders or as a lipid storage disorders, and are any one of several inborn errors of metabolism that result from enzyme defects affecting the ability of the body to oxidize fatty acids in order to produce energy within muscles, liver, and other cell types.
Mitochondrial trifunctional protein deficiency is an autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats to energy, particularly during periods without food. People with this disorder have inadequate levels of an enzyme that breaks down a certain group of fats called long-chain fatty acids.

Very long-chain specific acyl-CoA dehydrogenase, mitochondrial (VLCAD) is an enzyme that in humans is encoded by the ACADVL gene.

ACADM is a gene that provides instructions for making an enzyme called acyl-coenzyme A dehydrogenase that is important for breaking down (degrading) a certain group of fats called medium-chain fatty acids.

Malonyl-CoA decarboxylase deficiency (MCD) is an autosomal-recessive metabolic disorder caused by a genetic mutation that disrupts the activity of Malonyl-CoA decarboxylase. This enzyme breaks down Malonyl-CoA into acetyl-CoA and carbon dioxide.
Short-chain acyl-coenzyme A dehydrogenase deficiency (SCADD) is an autosomal recessive fatty acid oxidation disorder which affects enzymes required to break down a certain group of fats called short chain fatty acids.
Acyl-CoA dehydrogenases (ACADs) are a class of enzymes that function to catalyze the initial step in each cycle of fatty acid β-oxidation in the mitochondria of cells. Their action results in the introduction of a trans double-bond between C2 (α) and C3 (β) of the acyl-CoA thioester substrate. Flavin adenine dinucleotide (FAD) is a required co-factor in addition to the presence of an active site glutamate in order for the enzyme to function.

Acyl-CoA dehydrogenase, long chain is a protein that in humans is encoded by the ACADL gene.
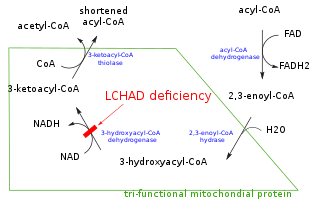
Mitochondrial trifunctional protein (MTP) is a protein attached to the inner mitochondrial membrane which catalyzes three out of the four steps in beta oxidation. MTP is a hetero-octamer composed of four alpha and four beta subunits:
Trifunctional enzyme subunit alpha, mitochondrial also known as hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase, alpha subunit is a protein that in humans is encoded by the HADHA gene. Mutations in HADHA have been associated with trifunctional protein deficiency or long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency.

Trifunctional enzyme subunit beta, mitochondrial (TP-beta) also known as 3-ketoacyl-CoA thiolase, acetyl-CoA acyltransferase, or beta-ketothiolase is an enzyme that in humans is encoded by the HADHB gene.

ACADSB is a human gene that encodes short/branched chain specific acyl-CoA dehydrogenase (SBCAD), an enzyme in the acyl CoA dehydrogenase family.

The human ETFA gene encodes the Electron-transfer-flavoprotein, alpha subunit, also known as ETF-α. Together with Electron-transfer-flavoprotein, beta subunit, encoded by the 'ETFB' gene, it forms the heterodimeric electron transfer flavoprotein (ETF). The native ETF protein contains one molecule of FAD and one molecule of AMP, respectively.

The human ETFB gene encodes the Electron-transfer-flavoprotein, beta subunit, also known as ETF-β. Together with Electron-transfer-flavoprotein, alpha subunit, encoded by the 'ETFA' gene, it forms the heterodimeric Electron transfer flavoprotein (ETF). The native ETF protein contains one molecule of FAD and one molecule of AMP, respectively.

Isobutyryl-CoA dehydrogenase, mitochondrial is an enzyme that in humans is encoded by the ACAD8 gene on chromosome 11.

Acyl-CoA dehydrogenase family member 9, mitochondrial is an enzyme that in humans is encoded by the ACAD9 gene. Mitochondrial Complex I Deficiency with varying clinical manifestations has been associated with mutations in ACAD9.
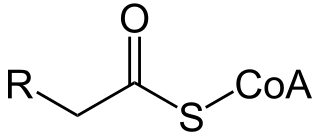
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.

Hydroxyacyl-Coenzyme A dehydrogenase (HADH) is an enzyme which in humans is encoded by the HADH gene.



