
Carnitine is a quaternary ammonium compound involved in metabolism in most mammals, plants, and some bacteria. In support of energy metabolism, carnitine transports long-chain fatty acids from the cytosol into mitochondria to be oxidized for free energy production, and also participates in removing products of metabolism from cells. Given its key metabolic roles, carnitine is concentrated in tissues like skeletal and cardiac muscle that metabolize fatty acids as an energy source. Generally individuals, including strict vegetarians, synthesize enough L-carnitine in vivo.
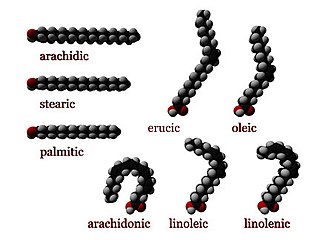
Numerous genetic disorders are caused by errors in fatty acid metabolism. These disorders may be described as fatty oxidation disorders or as a lipid storage disorders, and are any one of several inborn errors of metabolism that result from enzyme defects affecting the ability of the body to oxidize fatty acids in order to produce energy within muscles, liver, and other cell types.

Carnitine palmitoyltransferase I deficiency is a rare metabolic disorder that prevents the body from converting certain fats called long-chain fatty acids(LCFA) into energy, particularly during periods without food. It is caused by a mutation in CPT1A on chromosome 11.
Carnitine palmitoyltransferase II deficiency, sometimes shortened to CPT-II or CPT2, is an autosomal recessively inherited genetic metabolic disorder characterized by an enzymatic defect that prevents long-chain fatty acids from being transported into the mitochondria for utilization as an energy source. The disorder presents in one of three clinical forms: lethal neonatal, severe infantile hepatocardiomuscular and myopathic.
Fumarase deficiency is an exceedingly rare autosomal recessive metabolic disorder in the Krebs cycle, characterized by a deficiency of the enzyme fumarate hydratase, which causes a buildup of fumaric acid in the urine and a deficiency of malate. Only 13 cases were known worldwide in 1990, after which a cluster of 20 cases was documented in a community in Arizona, US that has practiced successive endogamy.

Palmitoylcarnitine is an ester derivative of carnitine involved in the metabolism of fatty acids. During the tricarboxylic acid cycle (TCA), fatty acids undergo a process known as β-oxidation to produce energy in the form of ATP. β-oxidation occurs primarily within mitochondria, however the mitochondrial membrane prevents the entry of long chain fatty acids (>C10), so the conversion of fatty acids such as palmitic acid is key. Palmitic acid is brought to the cell and once inside the cytoplasm is first converted to Palmitoyl-CoA. Palmitoyl-CoA has the ability to freely pass the outer mitochondrial membrane, but the inner membrane is impermeable to the Acyl-CoA and thioester forms of various long-chain fatty acids such as palmitic acid. The palmitoyl-CoA is then enzymatically transformed into palmitoylcarnitine via the Carnitine O-palmitoyltransferase family. The palmitoylcarnitine is then actively transferred into the inner membrane of the mitochondria via the carnitine-acylcarnitine translocase. Once inside the inner mitochondrial membrane, the same Carnitine O-palmitoyltransferase family is then responsible for transforming the palmitoylcarnitine back to the palmitoyl-CoA form.

Carnitine O-palmitoyltransferase is a mitochondrial transferase enzyme involved in the metabolism of palmitoylcarnitine into palmitoyl-CoA. A related transferase is carnitine acyltransferase.
Frataxin is a protein that in humans is encoded by the FXN gene.

Carnitine palmitoyltransferase I (CPT1) also known as carnitine acyltransferase I, CPTI, CAT1, CoA:carnitine acyl transferase (CCAT), or palmitoylCoA transferase I, is a mitochondrial enzyme responsible for the formation of acyl carnitines by catalyzing the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to l-carnitine. The product is often Palmitoylcarnitine, but other fatty acids may also be substrates. It is part of a family of enzymes called carnitine acyltransferases. This "preparation" allows for subsequent movement of the acyl carnitine from the cytosol into the intermembrane space of mitochondria.

Phosphorylase b kinase regulatory subunit alpha, liver isoform is an enzyme that in humans is encoded by the PHKA2 gene.

NAD-dependent malic enzyme, mitochondrial is a protein that in humans is encoded by the ME2 gene. This gene encodes a mitochondrial NAD-dependent malic enzyme, a homotetrameric protein, that catalyzes the oxidative decarboxylation of malate to pyruvate. It had previously been weakly linked to a syndrome known as Friedreich ataxia that has since been shown to be the result of mutation in a completely different gene.

NADP-dependent malic enzyme is a protein that in humans is encoded by the ME1 gene.
Trimethyllysine dioxygenase, mitochondrial is an enzyme that in humans is encoded by the TMLHE gene in chromosome X. Mutations in the TMLHE gene resulting in carnitine biosynthesis disruption have been associated with autism symptoms.

Elongation factor G 1, mitochondrial is a protein that in humans is encoded by the GFM1 gene. It is an EF-G homolog.
Elongation factor Ts, mitochondrial is a protein that in humans is encoded by the TSFM gene. It is an EF-Ts homolog.

Peroxisomal carnitine O-octanoyltransferase is an enzyme that in humans is encoded by the CROT gene.

Potassium voltage-gated channel, Shaw-related subfamily, member 3 also known as KCNC3 or Kv3.3 is a protein that in humans is encoded by the KCNC3.
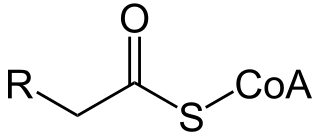
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.

AFG3 ATPase family gene 3-like 2 is a protein that in humans is encoded by the AFG3L2 gene.
Cytochrome c oxidase assembly factor COX20 is a protein that in humans is encoded by the COX20 gene. This gene encodes a protein that plays a role in the assembly of cytochrome c oxidase, an important component of the respiratory pathway. Mutations in this gene can cause mitochondrial complex IV deficiency. There are multiple pseudogenes for this gene. Alternative splicing results in multiple transcript variants.






