Related Research Articles

Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) is one of the family of mitochondrial diseases, which also include MERRF syndrome, and Leber's hereditary optic neuropathy. It was first characterized under this name in 1984. A feature of these diseases is that they are caused by defects in the mitochondrial genome which is inherited purely from the female parent.
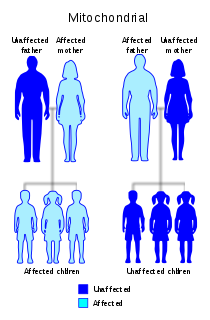
Diabetes and deafness (DAD) or maternally inherited diabetes and deafness (MIDD) or mitochondrial diabetes is a subtype of diabetes which is caused from a point mutation at position 3243 in human mitochondrial DNA, which consists of a circular genome. This affects the gene encoding tRNALeu. Because mitochondrial DNA is contributed to the embryo by the oocyte and not by spermatozoa, this disease is inherited from maternal family members only. As indicated by the name, MIDD is characterized by diabetes and sensorineural hearing loss.

Mitochondrially encoded 12S ribosomal RNA, also known as Mitochondrial-derived peptide MOTS-c or Mitochondrial open reading frame of the 12S rRNA-c is the SSU rRNA of the mitochondrial ribosome. In humans, 12S is encoded by the MT-RNR1 gene and is 959 nucleotides long. MT-RNR1 is one of the 37 genes contained in animal mitochondria genomes. Their 2 rRNA, 22 tRNA and 13 mRNA genes are very useful in phylogenetic studies, in particular the 12S and 16S rRNAs. The 12S rRNA is the mitochondrial homologue of the prokaryotic 16S and eukaryotic nuclear 18S ribosomal RNAs. Mutations in the MT-RNR1 gene may be associated with hearing loss.
MT-ATP6 is a mitochondrial gene with the full name 'mitochondrially encoded ATP synthase membrane subunit 6' that encodes the ATP synthase Fo subunit 6. This subunit belongs to the Fo complex of the large, transmembrane F-type ATP synthase. This enzyme, which is also known as complex V, is responsible for the final step of oxidative phosphorylation in the electron transport chain. Specifically, one segment of ATP synthase allows positively charged ions, called protons, to flow across a specialized membrane inside mitochondria. Another segment of the enzyme uses the energy created by this proton flow to convert a molecule called adenosine diphosphate (ADP) to ATP. Mutations in the MT-ATP6 gene have been found in approximately 10 to 20 percent of people with Leigh syndrome.
Mitochondrially encoded tRNA leucine 1 (UUA/G) also known as MT-TL1 is a transfer RNA which in humans is encoded by the mitochondrial MT-TL1 gene.
Mitochondrially encoded tRNA histidine, also known as MT-TH, is a transfer RNA which, in humans, is encoded by the mitochondrial MT-TH gene.
MT-ND5 is a gene of the mitochondrial genome coding for the NADH-ubiquinone oxidoreductase chain 5 protein (ND5). The ND5 protein is a subunit of NADH dehydrogenase (ubiquinone), which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variations in human MT-ND5 are associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) as well as some symptoms of Leigh's syndrome and Leber's hereditary optic neuropathy (LHON).
Cytochrome c oxidase subunit III (COX3) is an enzyme that in humans is encoded by the MT-CO3 gene. It is one of main transmembrane subunits of cytochrome c oxidase. Cytochrome c oxidase subunit III is also one of the three mitochondrial DNA (mtDNA) encoded subunits of respiratory complex IV. Variants of MT-CO3 have been associated with isolated myopathy, severe encephalomyopathy, Leber hereditary optic neuropathy, mitochondrial complex IV deficiency, and recurrent myoglobinuria.
Mitochondrially encoded tRNA valine also known as MT-TV is a transfer RNA which in humans is encoded by the mitochondrial MT-TV gene.
Mitochondrially encoded tRNA aspartic acid also known as MT-TD is a transfer RNA which in humans is encoded by the mitochondrial MT-TD gene.
Mitochondrially encoded tRNA phenylalanine also known as MT-TF is a transfer RNA which in humans is encoded by the mitochondrial MT-TF gene.
Mitochondrially encoded tRNA glycine also known as MT-TG is a transfer RNA which in humans is encoded by the mitochondrial MT-TG gene.
Mitochondrially encoded tRNA isoleucine also known as MT-TI is a transfer RNA which in humans is encoded by the mitochondrial MT-TI gene.
Mitochondrially encoded tRNA lysine also known as MT-TK is a transfer RNA which in humans is encoded by the mitochondrial MT-TK gene.
Mitochondrially encoded tRNA leucine 2 (CUN) also known as MT-TL2 is a transfer RNA which in humans is encoded by the mitochondrial MT-TL2 gene.
Mitochondrially encoded tRNA asparagine also known as MT-TN is a transfer RNA which in humans is encoded by the mitochondrial MT-TN gene.
Mitochondrially encoded tRNA arginine also known as MT-TR is a transfer RNA which in humans is encoded by the mitochondrial MT-TR gene.
Mitochondrially encoded tRNA threonine also known as MT-TT is a transfer RNA which in humans is encoded by the mitochondrial MT-TT gene.
Mitochondrially encoded tRNA tryptophan also known as MT-TW is a transfer RNA which in humans is encoded by the mitochondrial MT-TW gene.
Mitochondrially encoded tRNA tyrosine also known as MT-TY is a transfer RNA which in humans is encoded by the mitochondrial MT-TY gene.
References
- ↑ Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (April 1981). "Sequence and organization of the human mitochondrial genome". Nature. 290 (5806): 457–65. Bibcode:1981Natur.290..457A. doi:10.1038/290457a0. PMID 7219534. S2CID 4355527.
- 1 2 "MT-TE mitochondrially encoded tRNA glutamic acid [ Homo sapiens (human) ]". www.ncbi.nlm.nih.gov.
- ↑ "tRNA / transfer RNA | Learn Science at Scitable". www.nature.com.
- 1 2 3 4 5 "MT-TE gene". Genetics Home Reference. U.S. National Library of Medicine.
 This article incorporates text from this source, which is in the public domain .
This article incorporates text from this source, which is in the public domain . - ↑ Hanna MG, Nelson I, Sweeney MG, Cooper JM, Watkins PJ, Morgan-Hughes JA, Harding AE (May 1995). "Congenital encephalomyopathy and adult-onset myopathy and diabetes mellitus: different phenotypic associations of a new heteroplasmic mtDNA tRNA glutamic acid mutation". American Journal of Human Genetics. 56 (5): 1026–33. PMC 1801468 . PMID 7726155.
- ↑ Vialettes BH, Paquis-Flucklinger V, Pelissier JF, Bendahan D, Narbonne H, Silvestre-Aillaud P, Montfort MF, Righini-Chossegros M, Pouget J, Cozzone PJ, Desnuelle C (November 1997). "Phenotypic expression of diabetes secondary to a T14709C mutation of mitochondrial DNA. Comparison with MIDD syndrome (A3243G mutation): a case report". Diabetes Care. 20 (11): 1731–7. doi:10.2337/diacare.20.11.1731. PMID 9353617. S2CID 22589136.
- ↑ Reference, Genetics Home. "Cytochrome c oxidase deficiency". Genetics Home Reference.This article incorporates text from this source, which is in the public domain .
- ↑ Lax NZ, Gnanapavan S, Dowson SJ, Alston CL, He L, Polvikoski TM, Jaros E, O'Donovan DG, Yarham JW, Turnbull DM, Dean AF, Taylor RW (February 2013). "Early-onset cataracts, spastic paraparesis, and ataxia caused by a novel mitochondrial tRNAGlu (MT-TE) gene mutation causing severe complex I deficiency: a clinical, molecular, and neuropathologic study". Journal of Neuropathology and Experimental Neurology. 72 (2): 164–75. doi: 10.1097/NEN.0b013e31828129c5 . PMID 23334599.