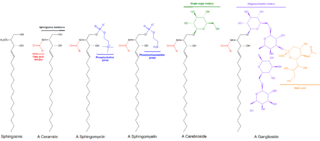
Sphingolipids are a class of lipids containing a backbone of sphingoid bases, a set of aliphatic amino alcohols that includes sphingosine. They were discovered in brain extracts in the 1870s and were named after the mythological sphinx because of their enigmatic nature. These compounds play important roles in signal transduction and cell recognition. Sphingolipidoses, or disorders of sphingolipid metabolism, have particular impact on neural tissue. A sphingolipid with an R group consisting of a hydrogen atom only is a ceramide. Other common R groups include phosphocholine, yielding a sphingomyelin, and various sugar monomers or dimers, yielding cerebrosides and globosides, respectively. Cerebrosides and globosides are collectively known as glycosphingolipids.

Carnitine palmitoyltransferase II deficiency is an autosomal recessively inherited genetic metabolic disorder characterized by an enzymatic defect that prevents long-chain fatty acids from being transported into the mitochondria for utilization as an energy source. The disorder presents in one of three clinical forms: lethal neonatal, severe infantile hepatocardiomuscular and myopathic.

Succinate dehydrogenase complex, subunit A, flavoprotein variant is a protein that in humans is encoded by the SDHA gene. This gene encodes a major catalytic subunit of succinate-ubiquinone oxidoreductase, a complex of the mitochondrial respiratory chain. The complex is composed of four nuclear-encoded subunits and is localized in the mitochondrial inner membrane. SDHA contains the FAD binding site where succinate is deprotonated and converted to fumarate. Mutations in this gene have been associated with a form of mitochondrial respiratory chain deficiency known as Leigh Syndrome. A pseudogene has been identified on chromosome 3q29. Alternatively spliced transcript variants encoding different isoforms have been found for this gene.

Carnitine palmitoyltransferase I (CPT1) also known as carnitine acyltransferase I, CPTI, CAT1, CoA:carnitine acyl transferase (CCAT), or palmitoylCoA transferase I, is a mitochondrial enzyme responsible for the formation of acyl carnitines by catalyzing the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to l-carnitine. The product is often Palmitoylcarnitine, but other fatty acids may also be substrates. It is part of a family of enzymes called carnitine acyltransferases. This "preparation" allows for subsequent movement of the acyl carnitine from the cytosol into the intermembrane space of mitochondria.

Trifunctional enzyme subunit beta, mitochondrial (TP-beta) also known as 3-ketoacyl-CoA thiolase, acetyl-CoA acyltransferase, or beta-ketothiolase is an enzyme that in humans is encoded by the HADHB gene.

MT-ND6 is a gene of the mitochondrial genome coding for the NADH-ubiquinone oxidoreductase chain 6 protein (ND6). The ND6 protein is a subunit of NADH dehydrogenase (ubiquinone), which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variations in the human MT-ND6 gene are associated with Leigh's syndrome, Leber's hereditary optic neuropathy (LHON) and dystonia.
MT-ND4L is a gene of the mitochondrial genome coding for the NADH-ubiquinone oxidoreductase chain 4L (ND4L) protein. The ND4L protein is a subunit of NADH dehydrogenase (ubiquinone), which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variants of human MT-ND4L are associated with increased BMI in adults and Leber's Hereditary Optic Neuropathy (LHON).
MT-ND1 is a gene of the mitochondrial genome coding for the NADH-ubiquinone oxidoreductase chain 1 (ND1) protein. The ND1 protein is a subunit of NADH dehydrogenase, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variants of the human MT-ND1 gene are associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Leigh's syndrome (LS), Leber's hereditary optic neuropathy (LHON) and increases in adult BMI.

In enzymology, a serine C-palmitoyltransferase (EC 2.3.1.50) is an enzyme that catalyzes the chemical reaction:

DNA polymerase subunit gamma is an enzyme that in humans is encoded by the POLG gene. Mitochondrial DNA polymerase is heterotrimeric, consisting of a homodimer of accessory subunits plus a catalytic subunit. The protein encoded by this gene is the catalytic subunit of mitochondrial DNA polymerase. Defects in this gene are a cause of progressive external ophthalmoplegia with mitochondrial DNA deletions 1 (PEOA1), sensory ataxic neuropathy dysarthria and ophthalmoparesis (SANDO), Alpers-Huttenlocher syndrome (AHS), and mitochondrial neurogastrointestinal encephalopathy syndrome (MNGIE).
Cytochrome c oxidase subunit III (COX3) is an enzyme that in humans is encoded by the MT-CO3 gene. It is one of main transmembrane subunits of cytochrome c oxidase. It is also one of the three mitochondrial DNA (mtDNA) encoded subunits of respiratory complex IV. Variants of it have been associated with isolated myopathy, severe encephalomyopathy, Leber hereditary optic neuropathy, mitochondrial complex IV deficiency, and recurrent myoglobinuria.

CDC34 is a gene that in humans encodes the protein Ubiquitin-conjugating enzyme E2 R1. This protein is a member of the ubiquitin-conjugating enzyme family, which catalyzes the covalent attachment of ubiquitin to other proteins.
Serine/threonine-protein phosphatase 2A 56 kDa regulatory subunit beta isoform is an enzyme that in humans is encoded by the PPP2R5B gene.

Serine palmitoyltransferase, long chain base subunit 2, also known as SPTLC2, is a protein which in humans is encoded by the SPTLC2 gene. SPTLC2 belongs to the class-II pyridoxal-phosphate-dependent aminotransferase family.

Palmitoyltransferase ZDHHC17 is an enzyme that contains a DHHC domain that in humans is encoded by the ZDHHC17 gene.

Neurofilament light polypeptide, also known as neurofilament light chain, is a neurofilament protein that in humans is encoded by the NEFL gene. Neurofilament light chain is a biomarker that can be measured with immunoassays in cerebrospinal fluid and plasma and reflects axonal damage in a wide variety of neurological disorders. It is a useful marker for disease monitoring in amyotrophic lateral sclerosis, multiple sclerosis, Alzheimer's disease, and more recently Huntington's disease. It is also promising marker for follow-up of patients with brain tumors. Higher numbers have been associated with increased mortality.
Hereditary sensory and autonomic neuropathy (HSAN) or hereditary sensory neuropathy (HSN) is a condition used to describe any of the types of this disease which inhibit sensation.
In molecular biology the DHHC domain is a protein domain that acts as an enzyme, which adds a palmitoyl chemical group to proteins in order to anchor them to cell membranes. The DHHC domain was discovered in 1999 and named after a conserved sequence motif found in its protein sequence. Roth and colleagues showed that the yeast Akr1p protein could palmitoylate Yck2p in vitro and inferred that the DHHC domain defined a large family of palmitoyltransferases. In mammals twenty three members of this family have been identified and their substrate specificities investigated. Some members of the family such as ZDHHC3 and ZDHHC7 enhance palmitoylation of proteins such as PSD-95, SNAP-25, GAP43, Gαs. Others such as ZDHHC9 showed specificity only toward the H-Ras protein. However, a recent study questions the involvement of classical enzyme-substrate recognition and specificity in the palmitoylation reaction. Several members of the family have been implicated in human diseases.
Hereditary sensory and autonomic neuropathy type I or hereditary sensory neuropathy type I is a group of autosomal dominant inherited neurological diseases that affect the peripheral nervous system particularly on the sensory and autonomic functions. The hallmark of the disease is the marked loss of pain and temperature sensation in the distal parts of the lower limbs. The autonomic disturbances, if present, manifest as sweating abnormalities.
The 1-deoxysphingolipids (1-deoxySLs) are an atypical and recently discovered class of sphingolipids (SLs). They are formed during the nove synthesis pathway and their essential C1-OH deficit causes the malfunctions of the following transformations to achieve complex sphingolipids. In general, sphingolipids are formed during a reaction that is catalyzed by the SPT enzyme (serine-palmitoyltransferase) where the condensation of serine and palmitoyl-CoA takes place. The origin of this rare sphingolipid, though, is due to a defect of the SPT which can also use alanine or glycine. This change is what forms the 1-deoxySL.



