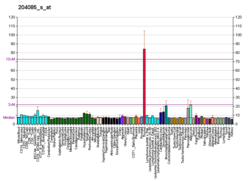
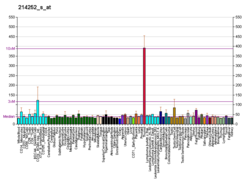
The neuronal ceroid lipofuscinoses (CLN or NCL) are a group of autosomal recessive, progressive encephalopathies in children. They are characterized by psychomotor deterioration, visual failure, and the accumulation of autofluorescent lipopigment in neurons and other cell types. The main childhood forms are the infantile type (Santavuori-Haltia disease; MIM 256730), the late infantile type (Jansky–Bielschowsky disease; MIM 204500), and the juvenile type (Batten disease; MIM 204200) based on the age of onset, clinical course, neurologic and ophthalmologic findings, and ultrastructural analysis (Carpenter et al., 1977 [PubMed 193610]).[supplied by OMIM][7]
A human clinical trial of gene therapy for the CLN5 form of Batten disease began in 2022 through the University of Rochester, using vectors developed by the Hughes research lab in New Zealand.[8]
Bessa C, Teixeira CA, Mangas M, etal. (2006). "Two novel CLN5 mutations in a Portuguese patient with vLINCL: insights into molecular mechanisms of CLN5 deficiency". Mol. Genet. Metab. 89 (3): 245–53. doi:10.1016/j.ymgme.2006.04.010. PMID16814585.
Cannelli N, Nardocci N, Cassandrini D, etal. (2007). "Revelation of a novel CLN5 mutation in early juvenile neuronal ceroid lipofuscinosis". Neuropediatrics. 38 (1): 46–9. doi:10.1055/s-2007-981449. PMID17607606. S2CID260241788.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.