Medium-chain acyl-CoA dehydrogenase deficiency is a disorder of fatty acid oxidation that impairs the body's ability to break down medium-chain fatty acids into acetyl-CoA. The disorder is characterized by hypoglycemia and sudden death without timely intervention, most often brought on by periods of fasting or vomiting.

Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency is a rare autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats into energy. This can become life-threatening, particularly during periods of fasting.

Isovaleric acidemia is a rare autosomal recessive metabolic disorder which disrupts or prevents normal metabolism of the branched-chain amino acid leucine. It is a classical type of organic acidemia.

Carnitine palmitoyltransferase I deficiency is a rare metabolic disorder that prevents the body from converting certain fats called long-chain fatty acids(LCFA) into energy, particularly during periods without food. It is caused by a mutation in CPT1A on chromosome 11.
Carnitine-acylcarnitine translocase deficiency is a rare, autosomal recessive metabolic disorder that prevents the body from converting long-chain fatty acids into energy, particularly during periods without food. Carnitine, a natural substance acquired mostly through the diet, is used by cells to process fats and produce energy. People with this disorder have a faulty enzyme that prevents long-chain fatty acids from being transported into the innermost part of the mitochondria for processing.
Mitochondrial trifunctional protein deficiency is an autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats to energy, particularly during periods without food.
Very long-chain acyl-coenzyme A dehydrogenase deficiency is a fatty-acid metabolism disorder which prevents the body from converting certain fats to energy, particularly during periods without food.

ACADM is a gene that provides instructions for making an enzyme called acyl-coenzyme A dehydrogenase that is important for breaking down (degrading) a certain group of fats called medium-chain fatty acids.
Glutaric acidemia type 2 is an autosomal recessive metabolic disorder that is characterised by defects in the ability of the body to use proteins and fats for energy. Incompletely processed proteins and fats can build up, leading to a dangerous chemical imbalance called acidosis. It is a metabolic myopathy, categorized under fatty acid metabolism disorder as that is the bioenergetic system that it affects the most. It also affects choline metabolism.

Malonic aciduria or malonyl-CoA decarboxylase deficiency (MCD) is an autosomal-recessive metabolic disorder caused by a genetic mutation that disrupts the activity of Malonyl-CoA decarboxylase. This enzyme breaks down Malonyl-CoA into acetyl-CoA and carbon dioxide.
2-Methylbutyryl-CoA dehydrogenase deficiency is an autosomal recessive metabolic disorder. It causes the body to be unable to process the amino acid isoleucine properly. Initial case reports identified individuals with developmental delay and epilepsy, however most cases identified through newborn screening have been asymptomatic.
Acyl-CoA dehydrogenases (ACADs) are a class of enzymes that function to catalyze the initial step in each cycle of fatty acid β-oxidation in the mitochondria of cells. Their action results in the introduction of a trans double-bond between C2 (α) and C3 (β) of the acyl-CoA thioester substrate. Flavin adenine dinucleotide (FAD) is a required co-factor in addition to the presence of an active site glutamate in order for the enzyme to function.

Acyl-CoA dehydrogenase, C-2 to C-3 short chain is an enzyme that in humans is encoded by the ACADS gene. This gene encodes a tetrameric mitochondrial flavoprotein, which is a member of the acyl-CoA dehydrogenase family. This enzyme catalyzes the initial step of the mitochondrial fatty acid beta-oxidation pathway. The ACADS gene is associated with short-chain acyl-coenzyme A dehydrogenase deficiency.
Isobutyryl-coenzyme A dehydrogenase deficiency is a rare metabolic disorder in which the body is unable to process certain amino acids properly.
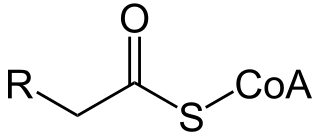
Acyl-CoA is a group of CoA-based coenzymes that metabolize carboxylic acids. Fatty acyl-CoA's are susceptible to beta oxidation, forming, ultimately, acetyl-CoA. The acetyl-CoA enters the citric acid cycle, eventually forming several equivalents of ATP. In this way, fats are converted to ATP, the common biochemical energy carrier.
3-hydroxyacyl-coenzyme A dehydrogenase deficiency is a rare condition that prevents the body from converting certain fats to energy, particularly during fasting. Normally, through a process called fatty acid oxidation, several enzymes work in a step-wise fashion to metabolize fats and convert them to energy. People with 3-hydroxyacyl-coenzyme A dehydrogenase deficiency have inadequate levels of an enzyme required for a step that metabolizes groups of fats called medium chain fatty acids and short chain fatty acids; for this reason this disorder is sometimes called medium- and short-chain 3-hydroxyacyl-coenzyme A dehydrogenase (M/SCHAD) deficiency.

ACADSB is a human gene that encodes short/branched chain specific acyl-CoA dehydrogenase (SBCAD), an enzyme in the acyl CoA dehydrogenase family.

Acyl-CoA dehydrogenase family member 9, mitochondrial is an enzyme that in humans is encoded by the ACAD9 gene. Mitochondrial Complex I Deficiency with varying clinical manifestations has been associated with mutations in ACAD9.
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.
Combined malonic and methylmalonic aciduria (CMAMMA), also called combined malonic and methylmalonic acidemia is an inherited metabolic disease characterized by elevated levels of malonic acid and methylmalonic acid. However, the methylmalonic acid levels exceed those of malonic acid. CMAMMA is not only an organic aciduria but also a defect of mitochondrial fatty acid synthesis (mtFASII). Some researchers have hypothesized that CMAMMA might be one of the most common forms of methylmalonic acidemia, and possibly one of the most common inborn errors of metabolism. Due to being infrequently diagnosed, it most often goes undetected.