Enoyl-CoA-(∆) isomerase (EC 5.3.3.8, also known as dodecenoyl-CoA- isomerase, 3,2-trans-enoyl-CoA isomerase, ∆3 ,∆2 -enoyl-CoA isomerase, or acetylene-allene isomerase, is an enzyme that catalyzes the conversion of cis- or trans-double bonds of coenzyme A bound fatty acids at gamma-carbon to trans double bonds at beta-carbon as below:

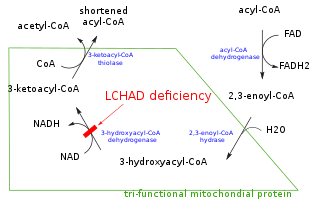
Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency is a rare autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats into energy. This can become life-threatening, particularly during periods of fasting.
In biochemistry and metabolism, beta oxidation (also β-oxidation) is the catabolic process by which fatty acid molecules are broken down in the cytosol in prokaryotes and in the mitochondria in eukaryotes to generate acetyl-CoA. Acetyl-CoA enters the citric acid cycle, generating NADH and FADH2, which are electron carriers used in the electron transport chain. It is named as such because the beta carbon of the fatty acid chain undergoes oxidation and is converted to a carbonyl group to start the cycle all over again. Beta-oxidation is primarily facilitated by the mitochondrial trifunctional protein, an enzyme complex associated with the inner mitochondrial membrane, although very long chain fatty acids are oxidized in peroxisomes.
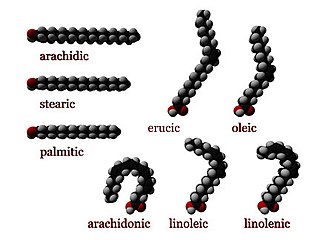
Numerous genetic disorders are caused by errors in fatty acid metabolism. These disorders may be described as fatty oxidation disorders or as a lipid storage disorders, and are any one of several inborn errors of metabolism that result from enzyme defects affecting the ability of the body to oxidize fatty acids in order to produce energy within muscles, liver, and other cell types.

Very long-chain specific acyl-CoA dehydrogenase, mitochondrial (VLCAD) is an enzyme that in humans is encoded by the ACADVL gene.
Chromosome 2 is one of the twenty-three pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 2 is the second-largest human chromosome, spanning more than 242 million base pairs and representing almost eight percent of the total DNA in human cells.

ACADM is a gene that provides instructions for making an enzyme called acyl-coenzyme A dehydrogenase that is important for breaking down (degrading) a certain group of fats called medium-chain fatty acids.

Short-chain acyl-coenzyme A dehydrogenase deficiency (SCADD) is an autosomal recessive fatty acid oxidation disorder which affects enzymes required to break down a certain group of fats called short chain fatty acids.

Acyl-CoA dehydrogenase, C-2 to C-3 short chain is an enzyme that in humans is encoded by the ACADS gene. This gene encodes a tetrameric mitochondrial flavoprotein, which is a member of the acyl-CoA dehydrogenase family. This enzyme catalyzes the initial step of the mitochondrial fatty acid beta-oxidation pathway. The ACADS gene is associated with short-chain acyl-coenzyme A dehydrogenase deficiency.
Mitochondrial trifunctional protein (MTP) is a protein attached to the inner mitochondrial membrane which catalyzes three out of the four steps in beta oxidation. MTP is a hetero-octamer composed of four alpha and four beta subunits:
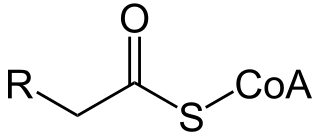
Acyl-CoA is a group of CoA-based coenzymes that metabolize carboxylic acids. Fatty acyl-CoA's are susceptible to beta oxidation, forming, ultimately, acetyl-CoA. The acetyl-CoA enters the citric acid cycle, eventually forming several equivalents of ATP. In this way, fats are converted to ATP, the common biochemical energy carrier.

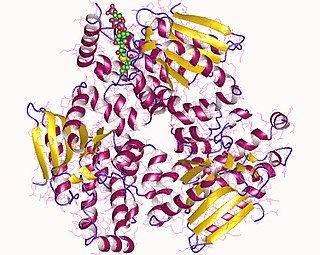
Trifunctional enzyme subunit alpha, mitochondrial also known as hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase, alpha subunit is a protein that in humans is encoded by the HADHA gene. Mutations in HADHA have been associated with trifunctional protein deficiency or long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency.
3-hydroxyacyl-coenzyme A dehydrogenase deficiency is a rare condition that prevents the body from converting certain fats to energy, particularly during fasting. Normally, through a process called fatty acid oxidation, several enzymes work in a step-wise fashion to metabolize fats and convert them to energy. People with 3-hydroxyacyl-coenzyme A dehydrogenase deficiency have inadequate levels of an enzyme required for a step that metabolizes groups of fats called medium chain fatty acids and short chain fatty acids; for this reason this disorder is sometimes called medium- and short-chain 3-hydroxyacyl-coenzyme A dehydrogenase (M/SCHAD) deficiency.

Trifunctional enzyme subunit beta, mitochondrial (TP-beta) also known as 3-ketoacyl-CoA thiolase, acetyl-CoA acyltransferase, or beta-ketothiolase is an enzyme that in humans is encoded by the HADHB gene.

2,4 Dienoyl-CoA reductase also known as DECR1 is an enzyme which in humans is encoded by the DECR1 gene which resides on chromosome 8. This enzyme catalyzes the following reactions
D-Bifunctional protein deficiency is an autosomal recessive peroxisomal fatty acid oxidation disorder. Peroxisomal disorders are usually caused by a combination of peroxisomal assembly defects or by deficiencies of specific peroxisomal enzymes. The peroxisome is an organelle in the cell similar to the lysosome that functions to detoxify the cell. Peroxisomes contain many different enzymes, such as catalase, and their main function is to neutralize free radicals and detoxify drugs. For this reason peroxisomes are ubiquitous in the liver and kidney. D-BP deficiency is the most severe peroxisomal disorder, often resembling Zellweger syndrome.

3-Methylglutaconyl-CoA hydratase, also known as MG-CoA hydratase and AUH, is an enzyme encoded by the AUH gene on chromosome 19. It is a member of the enoyl-CoA hydratase/isomerase superfamily, but it is the only member of that family that is able to bind to RNA. Not only does it bind to RNA, AUH has also been observed to be involved in the metabolic enzymatic activity, making it a dual-role protein. Mutations of this gene have been found to cause a disease called 3-Methylglutaconic Acuduria Type 1.

D-bifunctional protein (DBP), also known as peroxisomal multifunctional enzyme type 2 (MFP-2), as well as 17β-hydroxysteroid dehydrogenase type IV is a protein that in humans is encoded by the HSD17B4 gene. It's an alcohol oxidoreductase, specifically 17β-Hydroxysteroid dehydrogenase. It is involved in fatty acid β-oxidation and steroid metabolism.
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.

Hydroxyacyl-Coenzyme A dehydrogenase (HADH) is an enzyme which in humans is encoded by the HADH gene.