transcription factor – a protein that binds to DNA and regulates gene expression by promoting or suppressing transcription
promoter – a region of DNA that initiates transcription of a particular gene
Sigma factor – specialized bacterial co-factors that complex with RNA Polymerase and encode sequence specificity
coactivator – a protein that works with transcription factors to increase the rate of gene transcription
corepressor – a protein that works with transcription factors to decrease the rate of gene transcription
In molecular biology and genetics, transcriptional regulation is the means by which a cell regulates the conversion of DNA to RNA (transcription), thereby orchestrating gene activity. A single gene can be regulated in a range of ways, from altering the number of copies of RNA that are transcribed, to the temporal control of when the gene is transcribed. This control allows the cell or organism to respond to a variety of intra- and extracellular signals and thus mount a response. Some examples of this include producing the mRNA that encode enzymes to adapt to a change in a food source, producing the gene products involved in cell cycle specific activities, and producing the gene products responsible for cellular differentiation in multicellular eukaryotes, as studied in evolutionary developmental biology.
The regulation of transcription is a vital process in all living organisms. It is orchestrated by transcription factors and other proteins working in concert to finely tune the amount of RNA being produced through a variety of mechanisms. Bacteria and eukaryotes have very different strategies of accomplishing control over transcription, but some important features remain conserved between the two. Most importantly is the idea of combinatorial control, which is that any given gene is likely controlled by a specific combination of factors to control transcription. In a hypothetical example, the factors A and B might regulate a distinct set of genes from the combination of factors A and C. This combinatorial nature extends to complexes of far more than two proteins, and allows a very small subset (less than 10%) of the genome to control the transcriptional program of the entire cell.
In bacteria
The maltose operon is an example of a positive control of transcription. When maltose is not present in E. coli, no transcription of the maltose genes will occur, and there is no maltose to bind to the maltose activator protein. This prevents the activator protein from binding to the activator binding site on the gene, which in turn prevents RNA polymerase from binding to the maltose promoter. No transcription takes place.
Much of the early understanding of transcription came from bacteria,[2] although the extent and complexity of transcriptional regulation is greater in eukaryotes. Bacterial transcription is governed by three main sequence elements:
Promoters are elements of DNA that may bind RNA polymerase and other proteins for the successful initiation of transcription directly upstream of the gene.
Operators recognize repressor proteins that bind to a stretch of DNA and inhibit the transcription of the gene.
Positive control elements that bind to DNA and incite higher levels of transcription.[3]
While these means of transcriptional regulation also exist in eukaryotes, the transcriptional landscape is significantly more complicated both by the number of proteins involved as well as by the presence of introns and the packaging of DNA into histones.
The transcription of a basic bacterial gene is dependent on the strength of its promoter and the presence of activators or repressors. In the absence of other regulatory elements, a promoter's sequence-based affinity for RNA polymerases varies, which results in the production of different amounts of transcript. The variable affinity of RNA polymerase for different promoter sequences is related to regions of consensus sequence upstream of the transcription start site. The more nucleotides of a promoter that agree with the consensus sequence, the stronger the affinity of the promoter for RNA Polymerase likely is.[4]
When maltose is present in E. coli, it binds to the maltose activator protein (#1), which promotes maltose activator protein binding to the activator binding site (#2). This allows the RNA polymerase to bind to the mal promoter (#3). Transcription of malE, malF, and malG genes then proceeds (#4) as maltose activator protein and RNA polymerase moves down the DNA. malE encodes for maltose-binding periplasmic protein and helps maltose transport across the cell membrane. malF encodes for maltose transport system permease protein and helps translocate maltose across the cell membrane. malG encodes for transport system protein and also helps translocate maltose across the cell membrane.
In the absence of other regulatory elements, the default state of a bacterial transcript is to be in the "on" configuration, resulting in the production of some amount of transcript. This means that transcriptional regulation in the form of protein repressors and positive control elements can either increase or decrease transcription. Repressors often physically occupy the promoter location, occluding RNA polymerase from binding. Alternatively a repressor and polymerase may bind to the DNA at the same time with a physical interaction between the repressor preventing the opening of the DNA for access to the minus strand for transcription. This strategy of control is distinct from eukaryotic transcription, whose basal state is to be off and where co-factors required for transcription initiation are highly gene dependent.[8]
Sigma factors are specialized bacterial proteins that bind to RNA polymerases and orchestrate transcription initiation. Sigma factors act as mediators of sequence-specific transcription, such that a single sigma factor can be used for transcription of all housekeeping genes or a suite of genes the cell wishes to express in response to some external stimuli such as stress.[9]
In addition to processes that regulate transcription at the stage of initiation, mRNA synthesis is also controlled by the rate of transcription elongation.[10] RNA polymerase pauses occur frequently and are regulated by transcription factors, such as NusG and NusA, transcription-translation coupling, and mRNA secondary structure.[11][12]
The added complexity of generating a eukaryotic cell carries with it an increase in the complexity of transcriptional regulation. Eukaryotes have three RNA polymerases, known as Pol I, Pol II, and Pol III. Each polymerase has specific targets and activities, and is regulated by independent mechanisms. There are a number of additional mechanisms through which polymerase activity can be controlled. These mechanisms can be generally grouped into three main areas:
Control over polymerase access to the gene. This is perhaps the broadest of the three control mechanisms. This includes the functions of histone remodeling enzymes, transcription factors, enhancers and repressors, and many other complexes
Productive elongation of the RNA transcript. Once polymerase is bound to a promoter, it requires another set of factors to allow it to escape the promoter complex and begin successfully transcribing RNA.
Termination of the polymerase. A number of factors which have been found to control how and when termination occurs, which will dictate the fate of the RNA transcript.
All three of these systems work in concert to integrate signals from the cell and change the transcriptional program accordingly.
While in prokaryotic systems the basal transcription state can be thought of as nonrestrictive (that is, "on" in the absence of modifying factors), eukaryotes have a restrictive basal state which requires the recruitment of other factors in order to generate RNA transcripts. This difference is largely due to the compaction of the eukaryotic genome by winding DNA around histones to form higher order structures. This compaction makes the gene promoter inaccessible without the assistance of other factors in the nucleus, and thus chromatin structure is a common site of regulation. Similar to the sigma factors in prokaryotes, the general transcription factors (GTFs) are a set of factors in eukaryotes that are required for all transcription events. These factors are responsible for stabilizing binding interactions and opening the DNA helix to allow the RNA polymerase to access the template, but generally lack specificity for different promoter sites.[13] A large part of gene regulation occurs through transcription factors that either recruit or inhibit the binding of the general transcription machinery and/or the polymerase. This can be accomplished through close interactions with core promoter elements, or through the long distance enhancer elements.
Once a polymerase is successfully bound to a DNA template, it often requires the assistance of other proteins in order to leave the stable promoter complex and begin elongating the nascent RNA strand. This process is called promoter escape, and is another step at which regulatory elements can act to accelerate or slow the transcription process. Similarly, protein and nucleic acid factors can associate with the elongation complex and modulate the rate at which the polymerase moves along the DNA template.
At the level of chromatin state
In eukaryotes, genomic DNA is highly compacted in order to be able to fit it into the nucleus. This is accomplished by winding the DNA around protein octamers called histones, which has consequences for the physical accessibility of parts of the genome at any given time. Significant portions are silenced through histone modifications, and thus are inaccessible to the polymerases or their cofactors. The highest level of transcription regulation occurs through the rearrangement of histones in order to expose or sequester genes, because these processes have the ability to render entire regions of a chromosome inaccessible such as what occurs in imprinting.
Histone rearrangement is facilitated by post-translational modifications to the tails of the core histones. A wide variety of modifications can be made by enzymes such as the histone acetyltransferases (HATs), histone methyltransferases (HMTs), and histone deacetylases (HDACs), among others. These enzymes can add or remove covalent modifications such as methyl groups, acetyl groups, phosphates, and ubiquitin. Histone modifications serve to recruit other proteins which can either increase the compaction of the chromatin and sequester promoter elements, or to increase the spacing between histones and allow the association of transcription factors or polymerase on open DNA.[14] For example, H3K27 trimethylation by the polycomb complex PRC2 causes chromosomal compaction and gene silencing.[15] These histone modifications may be created by the cell, or inherited in an epigenetic fashion from a parent.
At the level of cytosine methylation
DNA methylation is the addition of a methyl group to the DNA that happens at cytosine. The image shows a cytosine single ring base and a methyl group added on to the 5 carbon. In mammals, DNA methylation occurs almost exclusively at a cytosine that is followed by a guanine.
Transcription regulation at about 60% of promoters is controlled by methylation of cytosines within CpG dinucleotides (where 5' cytosine is followed by 3' guanine or CpG sites). 5-methylcytosine (5-mC) is a methylated form of the DNA base cytosine (see Figure). 5-mC is an epigenetic marker found predominantly within CpG sites. About 28 million CpG dinucleotides occur in the human genome.[16] In most tissues of mammals, on average, 70% to 80% of CpG cytosines are methylated (forming 5-methylCpG or 5-mCpG).[17] Methylated cytosines within 5'cytosine-guanine 3' sequences often occur in groups, called CpG islands. About 60% of promoter sequences have a CpG island while only about 6% of enhancer sequences have a CpG island.[18] CpG islands constitute regulatory sequences, since if CpG islands are methylated in the promoter of a gene this can reduce or silence gene transcription.[19]
DNA methylation regulates gene transcription through interaction with methyl binding domain (MBD) proteins, such as MeCP2, MBD1 and MBD2. These MBD proteins bind most strongly to highly methylated CpG islands.[20] These MBD proteins have both a methyl-CpG-binding domain as well as a transcription repression domain.[20] They bind to methylated DNA and guide or direct protein complexes with chromatin remodeling and/or histone modifying activity to methylated CpG islands. MBD proteins generally repress local chromatin such as by catalyzing the introduction of repressive histone marks, or creating an overall repressive chromatin environment through nucleosome remodeling and chromatin reorganization.[20]
Transcription factors are proteins that bind to specific DNA sequences in order to regulate the expression of a gene. The binding sequence for a transcription factor in DNA is usually about 10 or 11 nucleotides long. As summarized in 2009, Vaquerizas et al. indicated there are approximately 1,400 different transcription factors encoded in the human genome by genes that constitute about 6% of all human protein encoding genes.[21] About 94% of transcription factor binding sites (TFBSs) that are associated with signal-responsive genes occur in enhancers while only about 6% of such TFBSs occur in promoters.[22]
EGR1 protein is a particular transcription factor that is important for regulation of methylation of CpG islands. An EGR1 transcription factor binding site is frequently located in enhancer or promoter sequences.[23] There are about 12,000 binding sites for EGR1 in the mammalian genome and about half of EGR1 binding sites are located in promoters and half in enhancers.[23] The binding of EGR1 to its target DNA binding site is insensitive to cytosine methylation in the DNA.[23]
While only small amounts of EGR1 transcription factor protein are detectable in cells that are un-stimulated, translation of the EGR1 gene into protein at one hour after stimulation is drastically elevated.[24] Expression of EGR1 transcription factor proteins, in various types of cells, can be stimulated by growth factors, neurotransmitters, hormones, stress and injury.[24] In the brain, when neurons are activated, EGR1 proteins are up-regulated and they bind to (recruit) the pre-existing TET1 enzymes which are highly expressed in neurons. TET enzymes can catalyse demethylation of 5-methylcytosine. When EGR1 transcription factors bring TET1 enzymes to EGR1 binding sites in promoters, the TET enzymes can demethylate the methylated CpG islands at those promoters. Upon demethylation, these promoters can then initiate transcription of their target genes. Hundreds of genes in neurons are differentially expressed after neuron activation through EGR1 recruitment of TET1 to methylated regulatory sequences in their promoters.[23]
The methylation of promoters is also altered in response to signals. The three mammalian DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) catalyze the addition of methyl groups to cytosines in DNA. While DNMT1 is a "maintenance" methyltransferase, DNMT3A and DNMT3B can carry out new methylations. There are also two spliceprotein isoforms produced from the DNMT3A gene: DNA methyltransferase proteins DNMT3A1 and DNMT3A2.[25]
The splice isoform DNMT3A2 behaves like the product of a classical immediate-early gene and, for instance, it is robustly and transiently produced after neuronal activation.[26] Where the DNA methyltransferase isoform DNMT3A2 binds and adds methyl groups to cytosines appears to be determined by histone post translational modifications.[27][28][29]
On the other hand, neural activation causes degradation of DNMT3A1 accompanied by reduced methylation of at least one evaluated targeted promoter.[30]
Through transcription factors and enhancers
Transcription factors
Transcription factors are proteins that bind to specific DNA sequences in order to regulate the expression of a given gene. There are approximately 1,400 transcription factors in the human genome and they constitute about 6% of all human protein coding genes.[21] The power of transcription factors resides in their ability to activate and/or repress wide repertoires of downstream target genes. The fact that these transcription factors work in a combinatorial fashion means that only a small subset of an organism's genome encodes transcription factors. Transcription factors function through a wide variety of mechanisms. In one mechanism, CpG methylation influences binding of most transcription factors to DNA—in some cases negatively and in others positively.[31] In addition, often they are at the end of a signal transduction pathway that functions to change something about the factor, like its subcellular localization or its activity. Post-translational modifications to transcription factors located in the cytosol can cause them to translocate to the nucleus where they can interact with their corresponding enhancers. Other transcription factors are already in the nucleus, and are modified to enable the interaction with partner transcription factors. Some post-translational modifications known to regulate the functional state of transcription factors are phosphorylation, acetylation, SUMOylation and ubiquitylation. Transcription factors can be divided in two main categories: activators and repressors. While activators can interact directly or indirectly with the core machinery of transcription through enhancer binding, repressors predominantly recruit co-repressor complexes leading to transcriptional repression by chromatin condensation of enhancer regions. It may also happen that a repressor may function by allosteric competition against a determined activator to repress gene expression: overlapping DNA-binding motifs for both activators and repressors induce a physical competition to occupy the site of binding. If the repressor has a higher affinity for its motif than the activator, transcription would be effectively blocked in the presence of the repressor. Tight regulatory control is achieved by the highly dynamic nature of transcription factors. Again, many different mechanisms exist to control whether a transcription factor is active. These mechanisms include control over protein localization or control over whether the protein can bind DNA.[32] An example of this is the protein HSF1, which remains bound to Hsp70 in the cytosol and is only translocated into the nucleus upon cellular stress such as heat shock. Thus the genes under the control of this transcription factor will remain untranscribed unless the cell is subjected to stress.[33]
Enhancers
Enhancers or cis-regulatory modules/elements (CRM/CRE) are non-coding DNA sequences containing multiple activator and repressor binding sites. Enhancers range from 200 bp to 1 kb in length and can be either proximal, 5' upstream to the promoter or within the first intron of the regulated gene, or distal, in introns of neighboring genes or intergenic regions far away from the locus. Through DNA looping, active enhancers contact the promoter dependently of the core DNA binding motif promoter specificity.[34] Promoter-enhancer dichotomy provides the basis for the functional interaction between transcription factors and transcriptional core machinery to trigger RNA Pol II escape from the promoter. Whereas one could think that there is a 1:1 enhancer-promoter ratio, studies of the human genome predict that an active promoter interacts with 4 to 5 enhancers. Similarly, enhancers can regulate more than one gene without linkage restriction and are said to "skip" neighboring genes to regulate more distant ones. Even though infrequent, transcriptional regulation can involve elements located in a chromosome different from one where the promoter resides. Proximal enhancers or promoters of neighboring genes can serve as platforms to recruit more distal elements.[35]
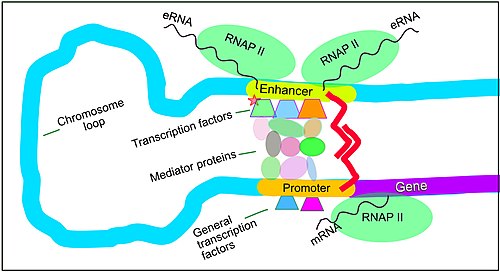
Enhance function in regulation of transcription in mammals. An active enhancer regulatory sequence of DNA is enabled to interact with the promoter DNA regulatory sequence of its target gene by formation of a chromosome loop. This can initiate messenger RNA (mRNA) synthesis by RNA polymerase II (RNAP II) bound to the promoter at the transcription start site of the gene. The loop is stabilized by one architectural protein anchored to the enhancer and one anchored to the promoter and these proteins are joined to form a dimer (red zigzags). Specific regulatory transcription factors bind to DNA sequence motifs on the enhancer. General transcription factors bind to the promoter. When a transcription factor is activated by a signal (here indicated as phosphorylation shown by a small red star on a transcription factor on the enhancer) the enhancer is activated and can now activate its target promoter. The active enhancer is transcribed on each strand of DNA in opposite directions by bound RNAP IIs. Mediator (a complex consisting of about 26 proteins in an interacting structure) communicates regulatory signals from the enhancer DNA-bound transcription factors to the promoter.
Up-regulated expression of genes in mammals can be initiated when signals are transmitted to the promoters associated with the genes. Cis-regulatory DNA sequences that are located in DNA regions distant from the promoters of genes can have very large effects on gene expression, with some genes undergoing up to 100-fold increased expression due to such a cis-regulatory sequence.[36] These cis-regulatory sequences include enhancers, silencers, insulators and tethering elements.[37] Among this constellation of sequences, enhancers and their associated transcription factor proteins have a leading role in the regulation of gene expression.[38]
Enhancers are sequences of the genome that are major gene-regulatory elements. Enhancers control cell-type-specific gene expression programs, most often by looping through long distances to come in physical proximity with the promoters of their target genes.[39] In a study of brain cortical neurons, 24,937 loops were found, bringing enhancers to promoters.[36] Multiple enhancers, each often at tens or hundred of thousands of nucleotides distant from their target genes, loop to their target gene promoters and coordinate with each other to control expression of their common target gene.[39]
The schematic illustration in this section shows an enhancer looping around to come into close physical proximity with the promoter of a target gene. The loop is stabilized by a dimer of a connector protein (e.g. dimer of CTCF or YY1), with one member of the dimer anchored to its binding motif on the enhancer and the other member anchored to its binding motif on the promoter (represented by the red zigzags in the illustration).[40] Several cell function specific transcription factor proteins (in 2018 Lambert et al. indicated there were about 1,600 transcription factors in a human cell[41]) generally bind to specific motifs on an enhancer[22] and a small combination of these enhancer-bound transcription factors, when brought close to a promoter by a DNA loop, govern the level of transcription of the target gene. Mediator (coactivator) (a complex usually consisting of about 26 proteins in an interacting structure) communicates regulatory signals from enhancer DNA-bound transcription factors directly to the RNA polymerase II (RNAP II) enzyme bound to the promoter.[42]
Enhancers, when active, are generally transcribed from both strands of DNA with RNA polymerases acting in two different directions, producing two eRNAs as illustrated in the Figure.[43] An inactive enhancer may be bound by an inactive transcription factor. Phosphorylation of the transcription factor may activate it and that activated transcription factor may then activate the enhancer to which it is bound (see small red star representing phosphorylation of a transcription factor bound to an enhancer in the illustration).[44] An activated enhancer begins transcription of its RNA before activating a promoter to initiate transcription of messenger RNA from its target gene.[45]
Typical enhancers are often of the size 151–240 base pairs.[46]
While enhancers are needed for transcription of a gene above a low level, clusters of enhancers, known as super-enhancers, can cause transcription of a target gene at a very high level.
A super-enhancer is a cluster of typical enhancers that drives a high level of transcription of a target gene
Super-enhancers are a group of typical enhancers, all located within a region of 10,000 to 60,000 nucleotides.[47][48] The typical enhancers within a super-enhancer simultaneously loop over from a distance to strongly increase initiation and transcription of a gene. The illustration in this section shows a super-enhancer of about 12,000 base pairs in length with four typical enhancers within its length. The enhancers are each associated with the same gene, transmitting signals from the transcription factors on each enhancer through a mediator protein complex to the promoter of the gene. Each typical enhancer within the cluster interacts with its own mediator multi-protein complex. The protein BRD4 complexes with each typical enhancer and stabilizes the super-enhancer structure.[49] Thus, there are a large number of proteins present in close association on a super-enhancer, including BRD4 proteins, transcription factors, 26 mediator proteins for each enhancer, etc.). Most of these proteins have a structured domain as well as a tail with an intrinsically disordered region.[50] The intrinsically disordered regions of these proteins interact with each other and usually form a water-excluding gel (phase-separated condensate) around the super-enhancer.[50]
As examples, in the case of the mouse α-globin super-enhancer, the five typical enhancers within the super-enhancer, acting together, increase transcription of the α-globin gene by 450-fold.[51] In the case of the mouse Wap super-enhancer, the three typical enhancers, acting together, increase transcription of the Wap gene by 1000-fold.[52]
In many types of cells, there are usually thousands of active typical enhancers and a few hundred super-enhancers. Super-enhancers (SEs) usually drive 2% to 4% of the actively transcribed regions of the genome. For instance, immune-system non-stimulated B cells have 140 super-enhancers (SEs) and 4,290 typical enhancers (TEs) (3.2% SEs).[53] Similarly, in mouse embryonic stem cells, there are 231 SEs compared to 8,794 TEs (2.6% SEs).[54] In neural stem cells there are 445 SEs and 9,436 TEs (4.7% SEs).[55]
While super-enhancers are only active at 2-4% of actively transcribed sites in a cell, they strongly recruit transcription machinery. The super-enhancers in a cell generally utilize 12% to 36% of the RNA polymerases, mediator proteins, BRD4 proteins, and other transcription machinery of the cell.[56]
Regulatory landscape
Transcriptional initiation, termination and regulation are mediated by "DNA looping" which brings together promoters, enhancers, transcription factors and RNA processing factors to accurately regulate gene expression.[57] Chromosome conformation capture (3C) and more recently Hi-C techniques provided evidence that active chromatin regions are "compacted" in nuclear domains or bodies where transcriptional regulation is enhanced.[58] The configuration of the genome is essential for enhancer-promoter proximity. Cell-fate decisions are mediated upon highly dynamic genomic reorganizations at interphase to modularly switch on or off entire gene regulatory networks through short to long range chromatin rearrangements.[59] Related studies demonstrate that metazoan genomes are partitioned in structural and functional units around a megabase long called Topological association domains (TADs) containing dozens of genes regulated by hundreds of enhancers distributed within large genomic regions containing only non-coding sequences. The function of TADs is to regroup enhancers and promoters interacting together within a single large functional domain instead of having them spread in different TADs.[60] However, studies of mouse development point out that two adjacent TADs may regulate the same gene cluster. The most relevant study on limb evolution shows that the TAD at the 5' of the HoxD gene cluster in tetrapod genomes drives its expression in the distal limb bud embryos, giving rise to the hand, while the one located at 3' side does it in the proximal limb bud, giving rise to the arm.[61] Still, it is not known whether TADs are an adaptive strategy to enhance regulatory interactions or an effect of the constrains on these same interactions. TAD boundaries are often composed by housekeeping genes, tRNAs, other highly expressed sequences and Short Interspersed Elements (SINE). While these genes may take advantage of their border position to be ubiquitously expressed, they are not directly linked with TAD edge formation. The specific molecules identified at boundaries of TADs are called insulators or architectural proteins because they not only block enhancer leaky expression but also ensure an accurate compartmentalization of cis-regulatory inputs to the targeted promoter. These insulators are DNA-binding proteins like CTCF and TFIIIC that help recruiting structural partners such as cohesins and condensins. The localization and binding of architectural proteins to their corresponding binding sites is regulated by post-translational modifications.[62] DNA binding motifs recognized by architectural proteins are either of high occupancy and at around a megabase of each other or of low occupancy and inside TADs. High occupancy sites are usually conserved and static while intra-TADs sites are dynamic according to the state of the cell therefore TADs themselves are compartmentalized in subdomains that can be called subTADs from few kb up to a TAD long (19). When architectural binding sites are at less than 100 kb from each other, Mediator proteins are the architectural proteins cooperate with cohesin. For subTADs larger than 100 kb and TAD boundaries, CTCF is the typical insulator found to interact with cohesion.[63]
Of the pre-initiation complex and promoter escape
In eukaryotes, ribosomal rRNA and the tRNAs involved in translation are controlled by RNA polymerase I (Pol I) and RNA polymerase III (Pol III) . RNA Polymerase II (Pol II) is responsible for the production of messenger RNA (mRNA) within the cell. Particularly for Pol II, much of the regulatory checkpoints in the transcription process occur in the assembly and escape of the pre-initiation complex. A gene-specific combination of transcription factors will recruit TFIID and/or TFIIA to the core promoter, followed by the association of TFIIB, creating a stable complex onto which the rest of the General Transcription Factors (GTFs) can assemble.[64] This complex is relatively stable, and can undergo multiple rounds of transcription initiation.[65] After the binding of TFIIB and TFIID, Pol II the rest of the GTFs can assemble. This assembly is marked by the post-translational modification (typically phosphorylation) of the C-terminal domain (CTD) of Pol II through a number of kinases.[66] The CTD is a large, unstructured domain extending from the RbpI subunit of Pol II, and consists of many repeats of the heptad sequence YSPTSPS. TFIIH, the helicase that remains associated with Pol II throughout transcription, also contains a subunit with kinase activity which will phosphorylate the serines 5 in the heptad sequence. Similarly, both CDK8 (a subunit of the massive multiprotein Mediator complex) and CDK9 (a subunit of the p-TEFb elongation factor), have kinase activity towards other residues on the CTD.[67] These phosphorylation events promote the transcription process and serve as sites of recruitment for mRNA processing machinery. All three of these kinases respond to upstream signals, and failure to phosphorylate the CTD can lead to a stalled polymerase at the promoter.
In vertebrates, the majority of gene promoters contain a CpG island with numerous CpG sites.[68] When many of a gene's promoter CpG sites are methylated the gene becomes silenced.[69] Colorectal cancers typically have 3 to 6 driver mutations and 33 to 66 hitchhiker or passenger mutations.[70] However, transcriptional silencing may be of more importance than mutation in causing progression to cancer. For example, in colorectal cancers about 600 to 800 genes are transcriptionally silenced by CpG island methylation (see regulation of transcription in cancer). Transcriptional repression in cancer can also occur by other epigenetic mechanisms, such as altered expression of microRNAs.[71] In breast cancer, transcriptional repression of BRCA1 may occur more frequently by over-expressed microRNA-182 than by hypermethylation of the BRCA1 promoter (see Low expression of BRCA1 in breast and ovarian cancers).
↑ Dukatz M, Holzer K, Choudalakis M, Emperle M, Lungu C, Bashtrykov P, Jeltsch A (December 2019). "H3K36me2/3 Binding and DNA Binding of the DNA Methyltransferase DNMT3A PWWP Domain Both Contribute to its Chromatin Interaction". J Mol Biol. 431 (24): 5063–5074. doi:10.1016/j.jmb.2019.09.006. PMID31634469. S2CID204832601.
↑ Voet, Donald Voet, Judith G. (2011). Biochemistry (4thed.). Hoboken, NJ: John Wiley & Sons. ISBN978-0-470-91745-9.{{cite book}}: CS1 maint: multiple names: authors list (link)
↑ Napolitano G, Lania L, Majello B (May 2014). "RNA polymerase II CTD modifications: how many tales from a single tail". J. Cell. Physiol. 229 (5): 538–44. doi:10.1002/jcp.24483. PMID24122273. S2CID44613555.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.