Types of non-coding DNA sequences
Noncoding genes
There are two types of genes: protein coding genes and noncoding genes. [14] Noncoding genes are an important part of non-coding DNA and they include genes for transfer RNA and ribosomal RNA. These genes were discovered in the 1960s. Prokaryotic genomes contain genes for a number of other noncoding RNAs but noncoding RNA genes are much more common in eukaryotes.
Typical classes of noncoding genes in eukaryotes include genes for small nuclear RNAs (snRNAs), small nucleolar RNAs (sno RNAs), microRNAs (miRNAs), short interfering RNAs (siRNAs), PIWI-interacting RNAs (piRNAs), and long noncoding RNAs (lncRNAs). In addition, there are a number of unique RNA genes that produce catalytic RNAs. [15]
Noncoding genes account for only a few percent of prokaryotic genomes [16] but they can represent a vastly higher fraction in eukaryotic genomes. [17] In humans, the noncoding genes take up at least 6% of the genome, largely because there are hundreds of copies of ribosomal RNA genes.[ citation needed ] Protein-coding genes occupy about 38% of the genome; a fraction that is much higher than the coding region because genes contain large introns.[ citation needed ]
The total number of noncoding genes in the human genome is controversial. Some scientists think that there are only about 5,000 noncoding genes while others believe that there may be more than 100,000 (see the article on Non-coding RNA). The difference is largely due to debate over the number of lncRNA genes. [18]
Promoters and regulatory elements
Promoters are DNA segments near the 5' end of the gene where transcription begins. They are the sites where RNA polymerase binds to initiate RNA synthesis. Every gene has a noncoding promoter.
Regulatory elements are sites that control the transcription of a nearby gene. They are almost always sequences where transcription factors bind to DNA and these transcription factors can either activate transcription (activators) or repress transcription (repressors). Regulatory elements were discovered in the 1960s and their general characteristics were worked out in the 1970s by studying specific transcription factors in bacteria and bacteriophage.[ citation needed ]
Promoters and regulatory sequences represent an abundant class of noncoding DNA but they mostly consist of a collection of relatively short sequences so they do not take up a very large fraction of the genome. The exact amount of regulatory DNA in mammalian genome is unclear because it is difficult to distinguish between spurious transcription factor binding sites and those that are functional. The binding characteristics of typical DNA-binding proteins were characterized in the 1970s and the biochemical properties of transcription factors predict that in cells with large genomes, the majority of binding sites will not be biologically functional.[ citation needed ]
Many regulatory sequences occur near promoters, usually upstream of the transcription start site of the gene. Some occur within a gene and a few are located downstream of the transcription termination site. In eukaryotes, there are some regulatory sequences that are located at a considerable distance from the promoter region. Some regulatory sequences are called enhancers or silencers but there is no rigorous definition that distinguishes them from other transcription factor binding sites. [19] [20] [21] [22]
Introns

Introns are the parts of a gene that are transcribed into the precursor RNA sequence, but ultimately removed by RNA splicing during the processing to mature RNA. Introns are found in both types of genes: protein-coding genes and noncoding genes. They are present in prokaryotes but they are much more common in eukaryotic genomes.[ citation needed ]
Group I and group II introns take up only a small percentage of the genome when they are present. Spliceosomal introns (see Figure) are only found in eukaryotes and they can represent a substantial proportion of the genome. In humans, for example, introns in protein-coding genes cover 37% of the genome. Combining that with about 1% coding sequences means that protein-coding genes occupy about 38% of the human genome. The calculations for noncoding genes are more complicated because there is considerable dispute over the total number of noncoding genes but taking only the well-defined examples means that noncoding genes occupy at least 6% of the genome. [23] [2]
Untranslated regions
The standard biochemistry and molecular biology textbooks describe non-coding nucleotides in mRNA located between the 5' end of the gene and the translation initiation codon. These regions are called 5'-untranslated regions or 5'-UTRs. Similar regions called 3'-untranslated regions (3'-UTRs) are found at the end of the gene. The 5'-UTRs and 3'UTRs are very short in bacteria but they can be several hundred nucleotides in length in eukaryotes. They contain short elements that control the initiation of translation (5'-UTRs) and transcription termination (3'-UTRs) as well as regulatory elements that may control mRNA stability, processing, and targeting to different regions of the cell. [24] [25] [26]
Origins of replication
DNA synthesis begins at specific sites called origins of replication. These are regions of the genome where the DNA replication machinery is assembled and the DNA is unwound to begin DNA synthesis. In most cases, replication proceeds in both directions from the replication origin.
The main features of replication origins are sequences where specific initiation proteins are bound. A typical replication origin covers about 100-200 base pairs of DNA. Prokaryotes have one origin of replication per chromosome or plasmid but there are usually multiple origins in eukaryotic chromosomes. The human genome contains about 100,000 origins of replication representing about 0.3% of the genome. [27] [28] [29]
Centromeres
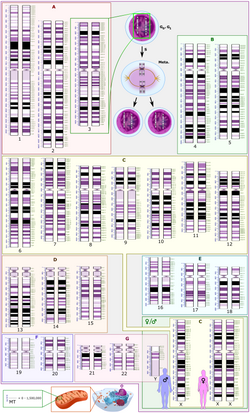
Centromeres are the sites where spindle fibers attach to newly replicated chromosomes in order to segregate them into daughter cells when the cell divides. Each eukaryotic chromosome has a single functional centromere that is seen as a constricted region in a condensed metaphase chromosome. Centromeric DNA consists of a number of repetitive DNA sequences that often take up a significant fraction of the genome because each centromere can be millions of base pairs in length. In humans, for example, the sequences of all 24 centromeres have been determined [31] and they account for about 6% of the genome. However, it is unlikely that all of this noncoding DNA is essential since there is considerable variation in the total amount of centromeric DNA in different individuals. [32] Centromeres are another example of functional noncoding DNA sequences that have been known for almost half a century and it is likely that they are more abundant than coding DNA.
Telomeres
Telomeres are regions of repetitive DNA at the end of a chromosome, which provide protection from chromosomal deterioration during DNA replication. Recent studies have shown that telomeres function to aid in its own stability. Telomeric repeat-containing RNA (TERRA) are transcripts derived from telomeres. TERRA has been shown to maintain telomerase activity and lengthen the ends of chromosomes. [33]
Scaffold attachment regions
Both prokaryotic and eukarotic genomes are organized into large loops of protein-bound DNA. In eukaryotes, the bases of the loops are called scaffold attachment regions (SARs) and they consist of stretches of DNA that bind an RNA/protein complex to stabilize the loop. There are about 100,000 loops in the human genome and each SAR consists of about 100 bp of DNA, so the total amount of DNA devoted to SARs accounts for about 0.3% of the human genome. [34]
Pseudogenes
Pseudogenes are mostly former genes that have become non-functional due to mutation, but the term also refers to inactive DNA sequences that are derived from RNAs produced by functional genes (processed pseudogenes). Pseudogenes are only a small fraction of noncoding DNA in prokaryotic genomes because they are eliminated by negative selection. In some eukaryotes, however, pseudogenes can accumulate because selection is not powerful enough to eliminate them (see Nearly neutral theory of molecular evolution).
The human genome contains about 15,000 pseudogenes derived from protein-coding genes and an unknown number derived from noncoding genes. [35] They may cover a substantial fraction of the genome (~5%) since many of them contain former intron sequences.
Pseudogenes are junk DNA by definition and they evolve at the neutral rate as expected for junk DNA. [36] Some former pseudogenes have secondarily acquired a function and this leads some scientists to speculate that most pseudogenes are not junk because they have a yet-to-be-discovered function. [37]
Repeat sequences, transposons and viral elements

Transposons and retrotransposons are mobile genetic elements. Retrotransposon repeated sequences, which include long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs), account for a large proportion of the genomic sequences in many species. Alu sequences, classified as a short interspersed nuclear element, are the most abundant mobile elements in the human genome. Some examples have been found of SINEs exerting transcriptional control of some protein-encoding genes. [38] [39] [40]
Endogenous retrovirus sequences are the product of reverse transcription of retrovirus genomes into the genomes of germ cells. Mutation within these retro-transcribed sequences can inactivate the viral genome. [41]
Over 8% of the human genome is made up of (mostly decayed) endogenous retrovirus sequences, as part of the over 42% fraction that is recognizably derived of retrotransposons, while another 3% can be identified to be the remains of DNA transposons. Much of the remaining half of the genome that is currently without an explained origin is expected to have found its origin in transposable elements that were active so long ago (> 200 million years) that random mutations have rendered them unrecognizable. [42] Genome size variation in at least two kinds of plants is mostly the result of retrotransposon sequences. [43] [44]
Highly repetitive DNA
Highly repetitive DNA consists of short stretches of DNA that are repeated many times in tandem (one after the other). The repeat segments are usually between 2 bp and 10 bp but longer ones are known. Highly repetitive DNA is rare in prokaryotes but common in eukaryotes, especially those with large genomes. It is sometimes called satellite DNA.
Most of the highly repetitive DNA is found in centromeres and telomeres (see above) and most of it is functional although some might be redundant. The other significant fraction resides in short tandem repeats (STRs; also called microsatellites) consisting of short stretches of a simple repeat such as ATC. There are about 350,000 STRs in the human genome and they are scattered throughout the genome with an average length of about 25 repeats. [45] [46]
Variations in the number of STR repeats can cause genetic diseases when they lie within a gene but most of these regions appear to be non-functional junk DNA where the number of repeats can vary considerably from individual to individual. This is why these length differences are used extensively in DNA fingerprinting.
Junk DNA
Junk DNA is DNA that has no biologically relevant function such as pseudogenes and fragments of once active transposons. Bacteria and viral genomes have very little junk DNA [47] [48] but some eukaryotic genomes may have a substantial amount of junk DNA. [49] The exact amount of nonfunctional DNA in humans and other species with large genomes has not been determined and there is considerable controversy in the scientific literature. [50] [51]
The nonfunctional DNA in bacterial genomes is mostly located in the intergenic fraction of non-coding DNA but in eukaryotic genomes it may also be found within introns. There are many examples of functional DNA elements in non-coding DNA, and it is erroneous to equate non-coding DNA with junk DNA.