Portion of gene's sequence which codes for protein
The coding region of a gene, also known as the coding DNA sequence (CDS), is the portion of a gene's DNA or RNA that codes for a protein.[1] Studying the length, composition, regulation, splicing, structures, and functions of coding regions compared to non-coding regions over different species and time periods can provide a significant amount of important information regarding gene organization and evolution of prokaryotes and eukaryotes.[2] This can further assist in mapping the human genome and developing gene therapy.[3]
Although this term is also sometimes used interchangeably with exon, it is not the exact same thing: the exon can be composed of the coding region as well as the 3' and 5' untranslated regions of the RNA, and so therefore, an exon would be partially made up of coding region. The 3' and 5' untranslated regions of the RNA, which do not code for protein, are termed non-coding regions and are not discussed on this page.[4]
There is often confusion between coding regions and exomes and there is a clear distinction between these terms. While the exome refers to all exons within a genome, the coding region refers to sections of the DNA (or primary transcript) or a singular section of processed mRNA which specifically codes for a certain kind of protein.
History
In 1978, Walter Gilbert published "Why Genes in Pieces" which first began to explore the idea that the gene is a mosaic—that each full nucleic acid strand is not coded continuously but is interrupted by "silent" non-coding regions. This was the first indication that there needed to be a distinction between the parts of the genome that code for protein, now called coding regions, and those that do not.[5]
Composition
Point mutation types: transitions (blue) are elevated compared to transversions (red) in GC-rich coding regions.
The evidence suggests that there is a general interdependence between base composition patterns and coding region availability.[6] The coding region is thought to contain a higher GC-content than non-coding regions. There is further research that discovered that the longer the coding strand, the higher the GC-content. Short coding strands are comparatively still GC-poor, similar to the low GC-content of the base composition translational stop codons like TAG, TAA, and TGA.[7]
GC-rich areas are also where the ratio point mutation type is altered slightly: there are more transitions, which are changes from purine to purine or pyrimidine to pyrimidine, compared to transversions, which are changes from purine to pyrimidine or pyrimidine to purine. The transitions are less likely to change the encoded amino acid and remain a silent mutation (especially if they occur in the third nucleotide of a codon) which is usually beneficial to the organism during translation and protein formation.[8]
This indicates that essential coding regions (gene-rich) are higher in GC-content and more stable and resistant to mutation compared to accessory and non-essential regions (gene-poor).[9] However, it is still unclear whether this came about through neutral and random mutation or through a pattern of selection.[10] There is also debate on whether the methods used, such as gene windows, to ascertain the relationship between GC-content and coding region are accurate and unbiased.[11]
Structure and function
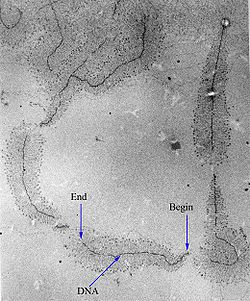
Transcription: RNA Polymerase (RNAP) uses a template DNA strand and begins coding at the promoter sequence (green) and ends at the terminator sequence (red) in order to encompass the entire coding region into the pre-mRNA (teal). The pre-mRNA is polymerised 5' to 3' and the template DNA read 3' to 5'An electron-micrograph of DNA strands decorated by hundreds of RNAP molecules too small to be resolved. Each RNAP is transcribing an RNA strand, which can be seen branching off from the DNA. "Begin" indicates the 3' end of the DNA, where RNAP initiates transcription; "End" indicates the 5' end, where the longer RNA molecules are completely transcribed.
In DNA, the coding region is flanked by the promoter sequence on the 5' end of the template strand and the termination sequence on the 3' end. During transcription, the RNA Polymerase (RNAP) binds to the promoter sequence and moves along the template strand to the coding region. RNAP then adds RNA nucleotides complementary to the coding region in order to form the mRNA, substituting uracil in place of thymine.[12] This continues until the RNAP reaches the termination sequence.[12]
After transcription and maturation, the mature mRNA formed encompasses multiple parts important for its eventual translation into protein. The coding region in an mRNA is flanked by the 5' untranslated region (5'-UTR) and 3' untranslated region (3'-UTR),[1] the 5' cap, and Poly-A tail. During translation, the ribosome facilitates the attachment of the tRNAs to the coding region, 3 nucleotides at a time (codons).[13] The tRNAs transfer their associated amino acids to the growing polypeptide chain, eventually forming the protein defined in the initial DNA coding region.
The coding region (teal) is flanked by untranslated regions, the 5' cap, and the poly(A) tail which together form the mature mRNA.
Regulation
The coding region can be modified in order to regulate gene expression.
Alkylation is one form of regulation of the coding region.[15] The gene that would have been transcribed can be silenced by targeting a specific sequence. The bases in this sequence would be blocked using alkyl groups, which create the silencing effect.[16]
While the regulation of gene expression manages the abundance of RNA or protein made in a cell, the regulation of these mechanisms can be controlled by a regulatory sequence found before the open reading frame begins in a strand of DNA. The regulatory sequence will then determine the location and time that expression will occur for a protein coding region.[17]
RNA splicing ultimately determines what part of the sequence becomes translated and expressed, and this process involves cutting out introns and putting together exons. Where the RNA spliceosome cuts, however, is guided by the recognition of splice sites, in particular the 5' splicing site, which is one of the substrates for the first step in splicing.[18] The coding regions are within the exons, which become covalently joined together to form the mature messenger RNA.
Mutations
Mutations in the coding region can have very diverse effects on the phenotype of the organism. While some mutations in this region of DNA/RNA can result in advantageous changes, others can be harmful and sometimes even lethal to an organism's survival. In contrast, changes in the non-coding region may not always result in detectable changes in phenotype.
Mutation types
Examples of the various forms of point mutations that may exist within coding regions. Such alterations may or may not have phenotypic changes, depending on whether or not they code for different amino acids during translation.
There are various forms of mutations that can occur in coding regions. One form is silent mutations, in which a change in nucleotides does not result in any change in amino acid after transcription and translation.[20] There also exist nonsense mutations, where base alterations in the coding region code for a premature stop codon, producing a shorter final protein. Point mutations, or single base pair changes in the coding region, that code for different amino acids during translation, are called missense mutations. Other types of mutations include frameshift mutations such as insertions or deletions.[20]
Formation
Some forms of mutations are hereditary (germline mutations), or passed on from a parent to its offspring.[21] Such mutated coding regions are present in all cells within the organism. Other forms of mutations are acquired (somatic mutations) during an organism's lifetime, and may not be constant cell-to-cell.[21] These changes can be caused by mutagens, carcinogens, or other environmental agents (ex. UV). Acquired mutations can also be a result of copy-errors during DNA replication and are not passed down to offspring. Changes in the coding region can also be de novo (new); such changes are thought to occur shortly after fertilization, resulting in a mutation present in the offspring's DNA while being absent in both the sperm and egg cells.[21]
Prevention
There exist multiple transcription and translation mechanisms to prevent lethality due to deleterious mutations in the coding region. Such measures include proofreading by some DNA Polymerases during replication, mismatch repair following replication,[22] and the 'Wobble Hypothesis' which describes the degeneracy of the third base within an mRNA codon.[23]
Constrained coding regions (CCRs)
While it is well known that the genome of one individual can have extensive differences when compared to the genome of another, recent research has found that some coding regions are highly constrained, or resistant to mutation, between individuals of the same species. This is similar to the concept of interspecies constraint in conserved sequences. Researchers termed these highly constrained sequences constrained coding regions (CCRs), and have also discovered that such regions may be involved in high purifying selection. On average, there is approximately 1 protein-altering mutation every 7 coding bases, but some CCRs can have over 100 bases in sequence with no observed protein-altering mutations, some without even synonymous mutations.[24] These patterns of constraint between genomes may provide clues to the sources of rare developmental diseases or potentially even embryonic lethality. Clinically validated variants and de novo mutations in CCRs have been previously linked to disorders such as infantile epileptic encephalopathy, developmental delay and severe heart disease.[24]
While identification of open reading frames within a DNA sequence is straightforward, identifying coding sequences is not, because the cell translates only a subset of all open reading frames to proteins.[26] Currently CDS prediction uses sampling and sequencing of mRNA from cells, although there is still the problem of determining which parts of a given mRNA are actually translated to protein. CDS prediction is a subset of gene prediction, the latter also including prediction of DNA sequences that code not only for protein but also for other functional elements such as RNA genes and regulatory sequences.
In both prokaryotes and eukaryotes, gene overlapping occurs relatively often in both DNA and RNA viruses as an evolutionary advantage to reduce genome size while retaining the ability to produce various proteins from the available coding regions.[27][28] For both DNA and RNA, pairwise alignments can detect overlapping coding regions, including short open reading frames in viruses, but would require a known coding strand to compare the potential overlapping coding strand with.[29] An alternative method using single genome sequences would not require multiple genome sequences to execute comparisons but would require at least 50 nucleotides overlapping in order to be sensitive.[30]
See also
Coding strand The DNA strand that codes for a protein
Exon The entire portion of the strand that is transcribed
Mature mRNA The portion of the mRNA transcription product that is translated
↑ Sakharkar MK, Chow VT, Kangueane P (2004). "Distributions of exons and introns in the human genome". In Silico Biology. 4 (4): 387–93. doi:10.3233/ISB-00142. PMID15217358.
↑ Peretó J. (2011) Wobble Hypothesis (Genetics). In: Gargaud M. et al. (eds) Encyclopedia of Astrobiology. Springer, Berlin, Heidelberg
1 2 Havrilla, J. M., Pedersen, B. S., Layer, R. M., & Quinlan, A. R. (2018). A map of constrained coding regions in the human genome. Nature Genetics, 88–95. doi:10.1101/220814
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.




{kind=link}
{kind=link}