Percentage of guanine and cytosine in DNA or RNA molecules
Nucleotide bonds showing AT and GC pairs. Arrows point to the hydrogen bonds.
In molecular biology and genetics, GC-content (or G+C content or guanine-cytosine content) is the percentage of nitrogenous bases in a DNA or RNA molecule that are either guanine (G) or cytosine (C).[1] This measure indicates the proportion of G and C bases out of an implied four total bases, also including adenine and thymine in DNA and adenine and uracil in RNA.
GC-content may be given for a certain fragment of DNA or RNA or for an entire genome. When it refers to a fragment, it may denote the GC-content of an individual gene or section of a gene (domain), a group of genes or gene clusters, a non-coding region, or a synthetic oligonucleotide such as a primer.
Structure
Qualitatively, guanine (G) and cytosine (C) undergo a specific hydrogen bonding with each other, whereas adenine (A) bonds specifically with thymine (T) in DNA and with uracil (U) in RNA. Quantitatively, each GC base pair is held together by three hydrogen bonds, while AT and AU base pairs are held together by two hydrogen bonds. To emphasize this difference, the base pairings are often represented as "G≡C" versus "A=T" or "A=U".
DNA with low GC-content is less stable than DNA with high GC-content; however, the hydrogen bonds themselves do not have a particularly significant impact on molecular stability, which is instead caused mainly by molecular interactions of base stacking.[2] Because of the thermostability of GC pairs, it was once presumed that high GC-content in DNA was a necessary adaptation to high temperatures, though this hypothesis was later refuted in 2001 by comparative analysis of over 100 prokaryotes.[3] Furthermore, P. putrefaciens, a species of bacteria with high GC-content DNA, has been observed to undergo autolysis more readily, thereby reducing the overall longevity of the cell.[4] Even so, it has been shown that there is a strong correlation between the optimal growth of prokaryotes at higher temperatures and the GC-content of structural RNAs, such as ribosomal RNA, transfer RNA, and many other non-coding RNAs.[3][5] The AU base pairs are less stable than the GC base pairs, making high-GC-content RNA structures more resistant to the effects of high temperatures.
More recently, it has been demonstrated that the most important factor contributing to the thermal stability of double-stranded nucleic acids is actually due to the base stackings of adjacent bases rather than the number of hydrogen bonds between the bases. There is more favorable stacking energy for GC pairs than for AT or AU pairs because of the relative positions of exocyclic groups. Additionally, there is a correlation between the order in which the bases stack and the thermal stability of the molecule as a whole.[6]
Determination
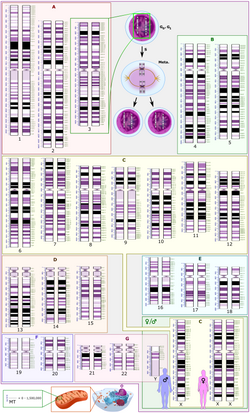
Schematic karyogram of a human, showing an overview of the human genome on G banding (which includes Giemsa-staining), wherein GC rich regions are lighter and GC poor regions are darker.
GC-content is usually expressed as a percentage value, but sometimes as a ratio (called G+C ratio or GC-ratio). GC-content percentage is calculated as[7]
The GC-content percentages as well as GC-ratio can be measured by several means, but one of the simplest methods is to measure the melting temperature of the DNA double helix using spectrophotometry. The absorbance of DNA at a wavelength of 260 nm increases fairly sharply when the double-stranded DNA molecule separates into two single strands when sufficiently heated.[9] The most commonly used protocol for determining GC-ratios uses flow cytometry for large numbers of samples.[10]
In an alternative manner, if the DNA or RNA molecule under investigation has been reliably sequenced, then GC-content can be accurately calculated by simple arithmetic or by using a variety of publicly available software tools, such as the free online GC calculatorArchived 26 February 2015 at the Wayback Machine .
Genomic content
Within-genome variation
The GC-ratio within a genome is found to be markedly variable. These variations in GC-ratio within the genomes of more complex organisms result in a mosaic-like formation with islet regions called isochores.[11] This results in the variations in staining intensity in chromosomes.[12] GC-rich isochores typically include many protein-coding genes within them, and thus determination of GC-ratios of these specific regions contributes to mapping gene-rich regions of the genome.[13][14]
Coding sequences
Within a long region of genomic sequence, genes are often characterised by having a higher GC-content in contrast to the background GC-content for the entire genome.[15] There is evidence that the length of the coding region of a gene is directly proportional to higher G+C content.[16] This has been pointed to the fact that the stop codon has a bias towards A and T nucleotides, and, thus, the shorter the sequence the higher the AT bias.[17]
Comparison of more than 1,000 orthologous genes in mammals showed marked within-genome variations of the third-codon position GC content, with a range from less than 30% to more than 80%.[18]
Among-genome variation
GC content is found to be variable with different organisms, the process of which is envisaged to be contributed to by variation in selection, mutational bias, and biased recombination-associated DNA repair.[19]
The average GC-content in human genomes ranges from 35% to 60% across 100-Kb fragments, with a mean of 41%.[20] The GC-content of Yeast (Saccharomyces cerevisiae) is 38%,[21] and that of another common model organism, thale cress (Arabidopsis thaliana), is 36%.[22] Because of the nature of the genetic code, it is virtually impossible for an organism to have a genome with a GC-content approaching either 0% or 100%. However, a species with an extremely low GC-content is Plasmodium falciparum (GC% = ~20%),[23] and it is usually common to refer to such examples as being AT-rich instead of GC-poor.[24]
Several mammalian species (e.g., shrew, microbat, tenrec, rabbit) have independently undergone a marked increase in the GC-content of their genes. These GC-content changes are correlated with species life-history traits (e.g., body mass or longevity) and genome size,[18] and might be linked to a molecular process called GC-biased gene conversion.[25]
Applications
Molecular biology
In polymerase chain reaction (PCR) experiments, the GC-content of short oligonucleotides known as primers is often used to predict their annealing temperature to the template DNA. A higher GC-content level indicates a relatively higher melting temperature.
Many sequencing technologies, such as Illumina sequencing, have trouble reading high-GC-content sequences. Bird genomes are known to have many such parts, causing the problem of "missing genes" expected to be present from evolution and phenotype but never sequenced — until improved methods were used.[26]
Systematics
The species problem in non-eukaryotic taxonomy has led to various suggestions in classifying bacteria, and the ad hoc committee on reconciliation of approaches to bacterial systematics of 1987 has recommended use of GC-ratios in higher-level hierarchical classification.[27] For example, the Actinomycetota are characterised as "high GC-content bacteria".[28] In Streptomyces coelicolor A3(2), GC-content is 72%.[29] With the use of more reliable, modern methods of molecular systematics, the GC-content definition of Actinomycetota has been abolished and low-GC bacteria of this clade have been found.[30]
Software tools
GCSpeciesSorter[31] and TopSort[32] are software tools for classifying species based on their GC-contents.
↑Levin RE, Van Sickle C (1976). "Autolysis of high-GC isolates of Pseudomonas putrefaciens". Antonie van Leeuwenhoek. 42 (1–2): 145–55. doi:10.1007/BF00399459. PMID7999. S2CID9960732.
↑Aïssani B, Bernardi G (October 1991). "CpG islands, genes and isochores in the genomes of vertebrates". Gene. 106 (2): 185–95. doi:10.1016/0378-1119(91)90198-K. PMID1937049.
↑Wuitschick JD, Karrer KM (1999). "Analysis of genomic G + C content, codon usage, initiator codon context and translation termination sites in Tetrahymena thermophila". J. Eukaryot. Microbiol. 46 (3): 239–47. doi:10.1111/j.1550-7408.1999.tb05120.x. PMID10377985. S2CID28836138.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.