
Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

Horizontal gene transfer (HGT) or lateral gene transfer (LGT) is the movement of genetic material between organisms other than by the ("vertical") transmission of DNA from parent to offspring (reproduction). HGT is an important factor in the evolution of many organisms. HGT is influencing scientific understanding of higher-order evolution while more significantly shifting perspectives on bacterial evolution.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.

Comparative genomics is a field of biological research in which the genomic features of different organisms are compared. The genomic features may include the DNA sequence, genes, gene order, regulatory sequences, and other genomic structural landmarks. In this branch of genomics, whole or large parts of genomes resulting from genome projects are compared to study basic biological similarities and differences as well as evolutionary relationships between organisms. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, comparative genomic approaches start with making some form of alignment of genome sequences and looking for orthologous sequences in the aligned genomes and checking to what extent those sequences are conserved. Based on these, genome and molecular evolution are inferred and this may in turn be put in the context of, for example, phenotypic evolution or population genetics.
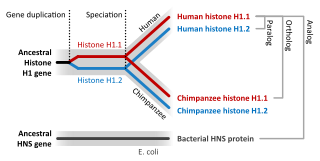
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).
Phylogenomics is the intersection of the fields of evolution and genomics. The term has been used in multiple ways to refer to analysis that involves genome data and evolutionary reconstructions. It is a group of techniques within the larger fields of phylogenetics and genomics. Phylogenomics draws information by comparing entire genomes, or at least large portions of genomes. Phylogenetics compares and analyzes the sequences of single genes, or a small number of genes, as well as many other types of data. Four major areas fall under phylogenomics:
In biology, a gene cassette is a type of mobile genetic element that contains a gene and a recombination site. Each cassette usually contains a single gene and tends to be very small; on the order of 500–1,000 base pairs. They may exist incorporated into an integron or freely as circular DNA. Gene cassettes can move around within an organism's genome or be transferred to another organism in the environment via horizontal gene transfer. These cassettes often carry antibiotic resistance genes. An example would be the kanMX cassette which confers kanamycin resistance upon bacteria.

Fiona Brinkman is a Professor in Bioinformatics and Genomics in the Department of Molecular Biology and Biochemistry at Simon Fraser University in British Columbia, Canada, and is a leader in the area of microbial bioinformatics. She is interested in developing "more sustainable, holistic approaches for infectious disease control and conservation of microbiomes".

The Z curve method is a bioinformatics algorithm for genome analysis. The Z-curve is a three-dimensional curve that constitutes a unique representation of a DNA sequence, i.e., for the Z-curve and the given DNA sequence each can be uniquely reconstructed from the other. The resulting curve has a zigzag shape, hence the name Z-curve.
GeneMark is a generic name for a family of ab initio gene prediction algorithms and software programs developed at the Georgia Institute of Technology in Atlanta. Developed in 1993, original GeneMark was used in 1995 as a primary gene prediction tool for annotation of the first completely sequenced bacterial genome of Haemophilus influenzae, and in 1996 for the first archaeal genome of Methanococcus jannaschii. The algorithm introduced inhomogeneous three-periodic Markov chain models of protein-coding DNA sequence that became standard in gene prediction as well as Bayesian approach to gene prediction in two DNA strands simultaneously. Species specific parameters of the models were estimated from training sets of sequences of known type. The major step of the algorithm computes for a given DNA fragment posterior probabilities of either being "protein-coding" in each of six possible reading frames or being "non-coding". The original GeneMark was an HMM-like algorithm; it could be viewed as approximation to known in the HMM theory posterior decoding algorithm for appropriately defined HMM model of DNA sequence.
Pathogenomics is a field which uses high-throughput screening technology and bioinformatics to study encoded microbe resistance, as well as virulence factors (VFs), which enable a microorganism to infect a host and possibly cause disease. This includes studying genomes of pathogens which cannot be cultured outside of a host. In the past, researchers and medical professionals found it difficult to study and understand pathogenic traits of infectious organisms. With newer technology, pathogen genomes can be identified and sequenced in a much shorter time and at a lower cost, thus improving the ability to diagnose, treat, and even predict and prevent pathogenic infections and disease. It has also allowed researchers to better understand genome evolution events - gene loss, gain, duplication, rearrangement - and how those events impact pathogen resistance and ability to cause disease. This influx of information has created a need for bioinformatics tools and databases to analyze and make the vast amounts of data accessible to researchers, and it has raised ethical questions about the wisdom of reconstructing previously extinct and deadly pathogens in order to better understand virulence.

In the fields of molecular biology and genetics, a pan-genome is the entire set of genes from all strains within a clade. More generally, it is the union of all the genomes of a clade. The pan-genome can be broken down into a "core pangenome" that contains genes present in all individuals, a "shell pangenome" that contains genes present in two or more strains, and a "cloud pangenome" that contains genes only found in a single strain. Some authors also refer to the cloud genome as "accessory genome" containing 'dispensable' genes present in a subset of the strains and strain-specific genes. Note that the use of the term 'dispensable' has been questioned, at least in plant genomes, as accessory genes play "an important role in genome evolution and in the complex interplay between the genome and the environment". The field of study of pangenomes is called pangenomics.

Horizontal gene transfer (HGT) refers to the transfer of genes between distant branches on the tree of life. In evolution, it can scramble the information needed to reconstruct the phylogeny of organisms, how they are related to one another.
The UCSC Genome Browser is an online and downloadable genome browser hosted by the University of California, Santa Cruz (UCSC). It is an interactive website offering access to genome sequence data from a variety of vertebrate and invertebrate species and major model organisms, integrated with a large collection of aligned annotations. The Browser is a graphical viewer optimized to support fast interactive performance and is an open-source, web-based tool suite built on top of a MySQL database for rapid visualization, examination, and querying of the data at many levels. The Genome Browser Database, browsing tools, downloadable data files, and documentation can all be found on the UCSC Genome Bioinformatics website.

In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
Microbial phylogenetics is the study of the manner in which various groups of microorganisms are genetically related. This helps to trace their evolution. To study these relationships biologists rely on comparative genomics, as physiology and comparative anatomy are not possible methods.
Horizontal or lateral gene transfer is the transmission of portions of genomic DNA between organisms through a process decoupled from vertical inheritance. In the presence of HGT events, different fragments of the genome are the result of different evolutionary histories. This can therefore complicate investigations of the evolutionary relatedness of lineages and species. Also, as HGT can bring into genomes radically different genotypes from distant lineages, or even new genes bearing new functions, it is a major source of phenotypic innovation and a mechanism of niche adaptation. For example, of particular relevance to human health is the lateral transfer of antibiotic resistance and pathogenicity determinants, leading to the emergence of pathogenic lineages.
PICRUSt is a bioinformatics software package. The name is an abbreviation for Phylogenetic Investigation of Communities by Reconstruction of Unobserved States.
Nikos Kyrpides is a Greek-American bioscientist who has worked on the origins of life, information processing, bioinformatics, microbiology, metagenomics and microbiome data science. He is a senior staff scientist at the Berkeley National Laboratory, head of the Prokaryote Super Program and leads the Microbiome Data Science program at the US Department of Energy Joint Genome Institute.

In phylogenetics, reconciliation is an approach to connect the history of two or more coevolving biological entities. The general idea of reconciliation is that a phylogenetic tree representing the evolution of an entity can be drawn within another phylogenetic tree representing an encompassing entity to reveal their interdependence and the evolutionary events that have marked their shared history. The development of reconciliation approaches started in the 1980s, mainly to depict the coevolution of a gene and a genome, and of a host and a symbiont, which can be mutualist, commensalist or parasitic. It has also been used for example to detect horizontal gene transfer, or understand the dynamics of genome evolution.