This article needs additional citations for verification .(March 2009) |
Low copy repeats (LCRs), also known as segmental duplications (SDs), are DNA sequences present in multiple locations within a genome that share high levels of sequence identity.
This article needs additional citations for verification .(March 2009) |
Low copy repeats (LCRs), also known as segmental duplications (SDs), are DNA sequences present in multiple locations within a genome that share high levels of sequence identity.
The repeats, or duplications, are typically 10–300 kb in length, and bear greater than 95% sequence identity. Though rare in most mammals, LCRs comprise a large portion of the human genome owing to a significant expansion during primate evolution. [1] In humans, chromosomes Y and 22 have the greatest proportion of SDs: 50.4% and 11.9% respectively. [2]
Misalignment of LCRs during non-allelic homologous recombination (NAHR) [3] is an important mechanism underlying the chromosomal microdeletion disorders as well as their reciprocal duplication partners. [4] Many LCRs are concentrated in "hotspots", such as the 17p11-12 region, 27% of which is composed of LCR sequence. NAHR and non-homologous end joining (NHEJ) within this region are responsible for a wide range of disorders, including Charcot–Marie–Tooth syndrome type 1A, [5] hereditary neuropathy with liability to pressure palsies, [5] Smith–Magenis syndrome, [6] and Potocki–Lupski syndrome. [3]
The two widely accepted methods for SD detection [7] are:

In genetics, a deletion is a mutation in which a part of a chromosome or a sequence of DNA is left out during DNA replication. Any number of nucleotides can be deleted, from a single base to an entire piece of chromosome. Some chromosomes have fragile spots where breaks occur, which result in the deletion of a part of the chromosome. The breaks can be induced by heat, viruses, radiation, or chemical reactions. When a chromosome breaks, if a part of it is deleted or lost, the missing piece of chromosome is referred to as a deletion or a deficiency.
Gene duplication is a major mechanism through which new genetic material is generated during molecular evolution. It can be defined as any duplication of a region of DNA that contains a gene. Gene duplications can arise as products of several types of errors in DNA replication and repair machinery as well as through fortuitous capture by selfish genetic elements. Common sources of gene duplications include ectopic recombination, retrotransposition event, aneuploidy, polyploidy, and replication slippage.
Gene conversion is the process by which one DNA sequence replaces a homologous sequence such that the sequences become identical after the conversion event. Gene conversion can be either allelic, meaning that one allele of the same gene replaces another allele, or ectopic, meaning that one paralogous DNA sequence converts another.

Haploinsufficiency in genetics describes a model of dominant gene action in diploid organisms, in which a single copy of the wild-type allele at a locus in heterozygous combination with a variant allele is insufficient to produce the wild-type phenotype. Haploinsufficiency may arise from a de novo or inherited loss-of-function mutation in the variant allele, such that it yields little or no gene product. Although the other, standard allele still produces the standard amount of product, the total product is insufficient to produce the standard phenotype. This heterozygous genotype may result in a non- or sub-standard, deleterious, and (or) disease phenotype. Haploinsufficiency is the standard explanation for dominant deleterious alleles.
Smith–Magenis Syndrome (SMS), also known as 17p- syndrome, is a microdeletion syndrome characterized by an abnormality in the short (p) arm of chromosome 17. It has features including intellectual disability, facial abnormalities, difficulty sleeping, and numerous behavioral problems such as self-harm. Smith–Magenis syndrome affects an estimated between 1 in 15,000 to 1 in 25,000 individuals.

Copy number variation (CNV) is a phenomenon in which sections of the genome are repeated and the number of repeats in the genome varies between individuals. Copy number variation is a type of structural variation: specifically, it is a type of duplication or deletion event that affects a considerable number of base pairs. Approximately two-thirds of the entire human genome may be composed of repeats and 4.8–9.5% of the human genome can be classified as copy number variations. In mammals, copy number variations play an important role in generating necessary variation in the population as well as disease phenotype.
Exon shuffling is a molecular mechanism for the formation of new genes. It is a process through which two or more exons from different genes can be brought together ectopically, or the same exon can be duplicated, to create a new exon-intron structure. There are different mechanisms through which exon shuffling occurs: transposon mediated exon shuffling, crossover during sexual recombination of parental genomes and illegitimate recombination.
The Olduvai domain, known until 2018 as DUF1220 and the NBPF repeat, is a protein domain that shows a striking human lineage-specific (HLS) increase in copy number and appears to be involved in human brain evolution. The protein domain has also been linked to several neurogenetic disorders such as schizophrenia and increased severity of autism. In 2018, it was named by its discoverers after Olduvai Gorge in Tanzania, one of the most important archaeological sites for early humans, to reflect data indicating its role in human brain size and evolution.

Leucine-rich repeat-containing protein 48 is a protein that in humans is encoded by the LRRC48 gene.
Potocki–Lupski syndrome (PTLS), also known as dup(17)p11.2p11.2 syndrome, trisomy 17p11.2 or duplication 17p11.2 syndrome, is a contiguous gene syndrome involving the microduplication of band 11.2 on the short arm of human chromosome 17 (17p11.2). The duplication was first described as a case study in 1996. In 2000, the first study of the disease was released, and in 2007, enough patients had been gathered to complete a comprehensive study and give it a detailed clinical description. PTLS is named for two researchers involved in the latter phases, Drs. Lorraine Potocki and James R. Lupski of Baylor College of Medicine.
8p23.1 duplication syndrome is a rare genetic disorder caused by a duplication of a region from human chromosome 8. This duplication syndrome has an estimated prevalence of 1 in 64,000 births and is the reciprocal of the 8p23.1 deletion syndrome. The 8p23.1 duplication is associated with a variable phenotype including one or more of speech delay, developmental delay, mild dysmorphism, with prominent forehead and arched eyebrows, and congenital heart disease (CHD).
Non-allelic homologous recombination (NAHR) is a form of homologous recombination that occurs between two lengths of DNA that have high sequence similarity, but are not alleles.
esophageal candidiasis1q21.1 deletion syndrome is a rare aberration of chromosome 1. A human cell has one pair of identical chromosomes on chromosome 1. With the 1q21.1 deletion syndrome, one chromosome of the pair is not complete, because a part of the sequence of the chromosome is missing. One chromosome has the normal length and the other is too short.
Genomic structural variation is the variation in structure of an organism's chromosome. It consists of many kinds of variation in the genome of one species, and usually includes microscopic and submicroscopic types, such as deletions, duplications, copy-number variants, insertions, inversions and translocations. Originally, a structure variation affects a sequence length about 1kb to 3Mb, which is larger than SNPs and smaller than chromosome abnormality. However, the operational range of structural variants has widened to include events > 50bp. The definition of structural variation does not imply anything about frequency or phenotypical effects. Many structural variants are associated with genetic diseases, however many are not. Recent research about SVs indicates that SVs are more difficult to detect than SNPs. Approximately 13% of the human genome is defined as structurally variant in the normal population, and there are at least 240 genes that exist as homozygous deletion polymorphisms in human populations, suggesting these genes are dispensable in humans. Rapidly accumulating evidence indicates that structural variations can comprise millions of nucleotides of heterogeneity within every genome, and are likely to make an important contribution to human diversity and disease susceptibility.

Unequal crossing over is a type of gene duplication or deletion event that deletes a sequence in one strand and replaces it with a duplication from its sister chromatid in mitosis or from its homologous chromosome during meiosis. It is a type of chromosomal crossover between homologous sequences that are not paired precisely. Normally genes are responsible for occurrence of crossing over. It exchanges sequences of different links between chromosomes. Along with gene conversion, it is believed to be the main driver for the generation of gene duplications and is a source of mutation in the genome.
A microdeletion syndrome is a syndrome caused by a chromosomal deletion smaller than 5 million base pairs spanning several genes that is too small to be detected by conventional cytogenetic methods or high resolution karyotyping. Detection is done by fluorescence in situ hybridization (FISH). Larger chromosomal deletion syndromes are detectable using karyotyping techniques.

Lorraine Potocki is a medical geneticist and educator at Texas Children's Hospital and is a professor at Baylor College of Medicine.
Segmental duplication are blocks of DNA ranging from 1 to 400 kb in length which recur at multiple sites within the genome, sharing greater than 90% similarity. Multiple studies have found a correlation between the location of segmental duplications and regions of chromosomal instability. This correlation suggests that they may be mediators of some genomic disorders. Segmental duplications are shown to be flanked on both sides by large homologous repeats, which exposes the region to recurrent rearrangement by nonallelic homologous recombination, leading to either deletion, duplication, or inversion of the original sequence.
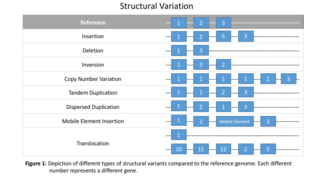
Structural variation in the human genome is operationally defined as genomic alterations, varying between individuals, that involve DNA segments larger than 1 kilo base (kb), and could be either microscopic or submicroscopic. This definition distinguishes them from smaller variants that are less than 1 kb in size such as short deletions, insertions, and single nucleotide variants.

Illegitimate recombination, or nonhomologous recombination, is the process by which two unrelated double stranded segments of DNA are joined. This insertion of genetic material which is not meant to be adjacent tends to lead to genes being broken causing the protein which they encode to not be properly expressed. One of the primary pathways by which this will occur is the repair mechanism known as non-homologous end joining (NHEJ).