
Jacobsen syndrome is a rare chromosomal disorder resulting from deletion of genes from chromosome 11 that includes band 11q24.1. It is a congenital disorder. Since the deletion takes place on the q arm of chromosome 11, it is also called 11q terminal deletion disorder. The deletion may range from 5 million to 16 million deleted DNA base pairs. The severity of symptoms depends on the number of deletions; the more deletions there are, the more severe the symptoms are likely to be.

22q13 deletion syndrome, also known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.
Potocki–Lupski syndrome (PTLS), also known as dup(17)p11.2p11.2 syndrome, trisomy 17p11.2 or duplication 17p11.2 syndrome, is a contiguous gene syndrome involving the microduplication of band 11.2 on the short arm of human chromosome 17 (17p11.2). The duplication was first described as a case study in 1996. In 2000, the first study of the disease was released, and in 2007, enough patients had been gathered to complete a comprehensive study and give it a detailed clinical description. PTLS is named for two researchers involved in the latter phases, Drs. Lorraine Potocki and James R. Lupski of Baylor College of Medicine.
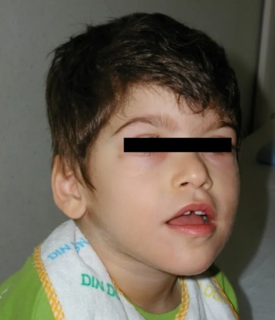
Pitt–Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, epilepsy, distinctive facial features, and possible intermittent hyperventilation followed by apnea. As more is learned about Pitt–Hopkins, the developmental spectrum of the disorder is widening, and can also include difficulties with anxiety, autism, ADHD, and sensory disorders. It is associated with an abnormality within chromosome 18; specifically, it is caused by an insufficient expression of the TCF4 gene.
3q29 microdeletion syndrome is a rare genetic disorder resulting from the deletion of a segment of chromosome 3. This syndrome was first described in 2005.
2q37 monosomy is a rare genetic disorder caused by a deletion of a segment at the end of chromosome 2.
22q11.2 duplication syndrome is a rare genetic disorder caused by a duplication of a segment at the end of chromosome 22.

Sotos syndrome is a rare genetic disorder characterized by excessive physical growth during the first years of life. Excessive growth often starts in infancy and continues into the early teen years. The disorder may be accompanied by autism, mild intellectual disability, delayed motor, cognitive, and social development, hypotonia, and speech impairments. Children with Sotos syndrome tend to be large at birth and are often taller, heavier, and have relatively large skulls (macrocephaly) than is normal for their age. Signs of the disorder, which vary among individuals, include a disproportionately large skull with a slightly protrusive forehead, large hands and feet, large mandible, hypertelorism, and downslanting eyes. Clumsiness, an awkward gait, and unusual aggressiveness or irritability may also occur.
Cognitive genomics is the sub-field of genomics pertaining to cognitive function in which the genes and non-coding sequences of an organism's genome related to the health and activity of the brain are studied. By applying comparative genomics, the genomes of multiple species are compared in order to identify genetic and phenotypical differences between species. Observed phenotypical characteristics related to the neurological function include behavior, personality, neuroanatomy, and neuropathology. The theory behind cognitive genomics is based on elements of genetics, evolutionary biology, molecular biology, cognitive psychology, behavioral psychology, and neurophysiology.

MECP2 duplication syndrome (M2DS) is a rare disease that is characterized by severe intellectual disability and impaired motor function. It is an X-linked genetic disorder caused by the overexpression of MeCP2 protein.
Burnside–Butler syndrome is a name that has been applied to the effects of microdeletion of DNA sequences involving four neurodevelopmental genes. Varying developmental and psychiatric disorders have been attributed to the microdeletion; however, the great majority of people with the deletion do not have any clinical features associated with it. More studies are needed to delineate the range of clinical presentation.
Birk-Barel syndrome is a rare genetic disorder associated with the KCNK9 gene. Signs and symptoms include mental retardation, hypotonia, hyperactivity, and syndromic facies.
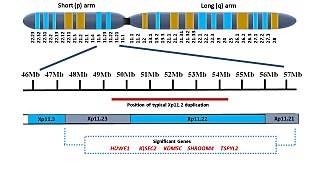
Xp11.2 duplication is a genomic variation marked by the duplication of an X chromosome region on the short arm p at position 11.2, defined by standard karyotyping (G-banding). This gene-rich, rearrangement prone region can be further divided into three loci - Xp11.21, Xp11.22 and Xp11.23. The duplication could involve any combination of these three loci. While the length of the duplication can vary from 0.5Mb to 55 Mb, most duplications measure about 4.5Mb and typically occur in the region of 11.22-11.23. Most affected females show preferential activation of the duplicated X chromosome. Features of affected individuals vary significantly, even among members of the same family. The Xp11.2 duplication can be 'silent' - presenting no obvious symptoms in carriers - which is known from the asymptomatic parents of affected children carrying the duplication. The common symptoms include intellectual disabilities, speech delay and learning difficulties, while in rare cases, children have seizures and a recognizable brain wave pattern when assessed by EEG (electroencephalography).

Ring chromosome 15 is a condition that arises when chromosome 15 fuses to form a ring chromosome. Usually, ring chromosome 15 forms due to the modification or deletion of genetic information on chromosome 15 in the preliminary stages of embryonic development, but it can rarely also be inherited.
SYNGAP1-related intellectual disability is a monogenetic developmental and epileptic encephalopathy that affects the central nervous system. Symptoms include intellectual disability, epilepsy, autism, sensory processing deficits, hypotonia and unstable gait.

17q12 microdeletion syndrome, also known as 17q12 deletion syndrome, is a rare chromosomal anomaly caused by the deletion of a small amount of material from a region in the long arm of chromosome 17. It is typified by deletion of the HNF1B gene, resulting in kidney abnormalities and renal cysts and diabetes syndrome. It also has neurocognitive effects, and has been implicated as a genetic factor for autism and schizophrenia.
XYYYY syndrome, also known as 49,XYYYY, is an exceptionally rare chromosomal disorder in which a male has three additional copies of the Y chromosome. Only seven non-mosaic cases of the disorder have ever been recorded in the medical literature, as well as five mosaic cases, of which two had more 48,XYYY than 49,XYYYY cells. Due to the extreme rarity of the disorder, little is understood about it, and the phenotype appears to be variable.
CHAMP1-associated intellectual disability syndrome, also known as autosomal dominant intellectual disability type 40 is a rare genetic disorder which is characterized by intellectual disabilities, developmental delays, facial dysmorphisms, and other anomalies.
