Mantle cell lymphoma (MCL) is a type of non-Hodgkin's lymphoma, comprising about 6% of cases.[1][2] It is named for the mantle zone of the lymph nodes where it develops.[3][4] The term 'mantle cell lymphoma' was first adopted by Raffeld and Jaffe in 1991.[5]
Lymph nodes of the head and neck, from Gray's Anatomy (click image to enlarge)
Signs and symptoms
People with mantle cell lymphoma typically present with symptoms later in life, with a median age of onset between 60 and 70 years of age.[8] In Western countries MCL accounts for around 7% of adult non-Hodgkin's lymphomas, with between 4 and 8 per cases per million diagnosed each year. The incidence of MCL increases with age. In the United States, the median age for its diagnosis is 68 years. Three-quarters of patients are men. In addition, patients are more likely to be Caucasian.[5]
People commonly present with a non-localizing lymphadenopathy (enlarged lymph nodes) with B symptoms including fevers, chills and night sweats sometimes being present.[8] 80% of patients present with stage 3 or 4 disease (advanced disease) at the time of diagnosis, with involvement of the bone marrow, liver or gastrointestinal tract.[8][9] 25% of patients present with a bulky lymphadenopathy characterized by lymph nodes greater than 10cm in size.[8] Other patients may present with central nervous system (CNS) involvement, which is associated with a very poor prognosis.[8] However, CNS involvement is rare at diagnosis.[10]
A rare subtype, known as non-nodal mantle cell lymphoma, presents without lymph node swelling (non-nodal) with circulating lymphoma cells (leukemic presentation).[11] This type of mantle cell lymphoma is associated with a more indolent, asymptomatic and slowly progressive course, however malignant transformation to aggressive forms is possible.[12][8]
Mantle cell lymphoma has been reported in rare cases to be associated with severe allergic reactions to mosquito bites. These reactions involve extensive allergic reactions to mosquito bites which range from greatly enlarged bite sites that may be painful and involve necrosis to systemic symptoms (e.g. fever, swollen lymph nodes, abdominal pain, and diarrhea), or, in extremely rare cases, to life-threatening anaphylaxis. In several of these cases, the mosquito bite allergy reaction occurred prior to the diagnosis of MCL suggesting that mosquito bite allergy can be a manifestation of early-developing mantle cell lymphoma.[13][14]
Pathogenesis
Histology of a normal lymphoid follicle, with yellow arrows pointing at mantle zone.
MCL, like most cancers, results from the acquisition of a combination of (non-inherited) genetic mutations in somatic cells. This leads to a clonal expansion of malignant B lymphocytes. The factors that initiate the genetic alterations are typically not identifiable, and usually occur in people with no particular risk factors for lymphoma development. Because it is an acquired genetic disorder, MCL is neither communicable [15] nor inheritable.[16]
A defining characteristic of MCL is mutation and overexpression of cyclin D1, a cell cycle gene, that contributes to the abnormal proliferation of the malignant cells. MCL cells may also be resistant to drug-induced apoptosis, making them harder to cure with chemotherapy or radiation. Cells affected by MCL proliferate in a nodular or diffuse pattern with two main cytologic variants, typical or blastic. Typical cases are small to intermediate-sized cells with irregular nuclei. Blastic (aka blastoid) variants have intermediate to large-sized cells with finely dispersed chromatin, and are more aggressive in nature.[17] The tumor cells accumulate in the lymphoid system, including lymph nodes and the spleen, with non-useful cells eventually rendering the system dysfunctional. MCL may also replace normal cells in the bone marrow, which impairs normal blood cell production.[18]
Diagnosis
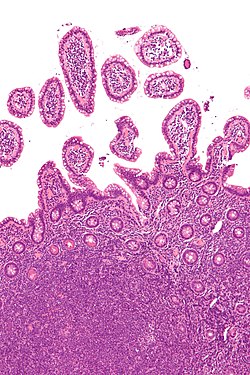
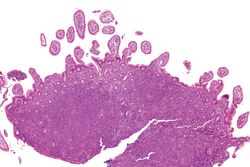
Lymph node with mantle cell lymphoma (low power view, H&E)Mantle cell lymphoma. Notice the irregular nuclear contours of the medium-sized lymphoma cells and the presence of a pink histiocyte. By immunohistochemistry the lymphoma cells expressed CD20, CD5 and cyclin D1 (high power view, H&E)Micrograph of terminal ileum with mantle cell lymphoma (bottom of image). H&E stain.Micrograph of terminal ileum with mantle cell lymphoma (bottom of image - brown colour). Cyclin D1immunostain.
The history and physical examination may reveal some of the signs and symptoms consistent with Mantle Cell Lymphoma. Biopsy of the involved tissues (such as the lymph nodes, bone marrow, gastrointestinal tract, spleen or other areas) shows the characteristic histopathologic changes of MCL. There are distinct growth patterns of MCL seen on biopsy; these include the diffuse type, nodular type, mantle zone lymphoma and in situ mantle cell lymphoma.[8] In the diffuse growth pattern, there is a diffuse growth of lymphoma cells throughout the lymph node resulting in effacement of the architecture of the lymph node.[8] In the nodular type, there are large nodules of MCL cells in the lymph node with no germinal centers observed.[8] In MCL with expansion of the mantle zone, the lymphoma cells cause expansion of the mantle zone around normal germinal centers.[8] And in MCL in situ, the lymphoma cells are contained within the mantle zone without expansion.[8] Histologically, the lymphoma cells in classic MCL are characterized as small to medium lymphocytes with scant cytoplasm and clumped chromatin with prominent nuclear clefts and the nucleoli are not visible.[8] There are cytologic subtypes; the blastoid subtype, is characterized by round nuclei, fine chromatin with some distinct nucleoli.[8] The pleomorphic subtype is characterized by nuclei that vary in size and shape with some having a cleaved form.[8] The blastoid and pleomorphic subtypes of MCL are associated with a more aggressive course.[8]
The most common B-cell type seen in MCL is a pre-germinal center cell (that has not yet undergone the germinal center reaction), that is CD5, CD20, CD19 positive with expression of IgM and IgD with monoclonal kappa and gamma light chains.[19]CD23 and CD200 are usually negative and cyclin-D1 (a cell cycle regulatory protein controlling transition from the G1 phase to the S phase in the cell cycle) is classically overexpressed in MCL.[19]SOX11 (a transcription factor controlling genes involved in cell survival) is characteristically over-expressed in MCL as well.[19]Ki-67, a marker of cell proliferation, if elevated (greater than 30% expression) is associated with an aggressive course of MCL.[19]
MRI of the brain and the spine are performed in cases of MCL with suspected central nervous system involvement.[19] And, since 40-80% of MCL presents with gastrointestinal involvement at the time of diagnosis, endoscopy (colonoscopy and esophagogastroduodenoscopy (EGD)) with biopsies may also aid in the diagnosis, but they are not always required for the diagnosis of MCL.[19]
The diagnosis may be complicated as a minority of cases of multiple myeloma, chronic lymphocytic leukemia and plasma cell leukemia may also present with the t(11;14)(q13;q32) translocation.[19] The diagnosis may be complicated further as some cases of MCL present atypically; these rare subtypes include CD10-positive MCL, CD5-negative MCL, cyclin D1-negative MCL, CD200-positive MCL, SOX-11-negative MCL, and CD23-positive MCL.[19] The cyclin-D1-negative MCL subtypes usually result in lymphomagenesis via over-expression of cyclin D2, cyclin D3 or cyclin E, which also lead to cell cycle hyperactivity and have a similar prognosis to the main cyclin-D1 variant of MCL.[8]
The Lugano and Ann Arbor Staging systems are two commonly used clinical staging criteria used to stage the disease, allowing decisions to be made with respect to treatment, prognosis and salvage therapy.[19]
Treatments
There are no proven standards of treatment for MCL, and there is no consensus among specialists on how to treat it optimally.[20] Many regimens are available and often get good response rates, but patients almost always get disease progression after chemotherapy. Each relapse is typically more difficult to treat, and relapse is generally faster. As of 2023 it is incurable though some patients can live many years after their initial diagnosis.[5] Regimens are available that treat relapses, and new approaches are under test. Because of the aforementioned factors, many MCL patients enroll in clinical trials to get the latest treatments – a survey at a specialist treatment centre in the UK showed that In total 58·7% of patients treated at the hospital were enrolled on at least one clinical trial.[21] Indeed, this might well be a recommendation by the patient's care team in the hope it will give them access to the latest advances.[10]
There are four classes of treatments in general use: chemotherapy, immunotherapy, radioimmunotherapy and biologic agents. The phases of treatment are generally: frontline, following diagnosis, consolidation, after frontline response (to prolong remissions), and relapse. Relapse is usually experienced multiple times.[22]
Chemotherapy
Chemotherapy is widely used as frontline treatment, and often is not repeated in relapse due to side effects. Alternate chemotherapy is sometimes used at first relapse. For frontline treatment, CHOP with rituximab is the most common chemotherapy, and often given as outpatient by IV. A stronger chemotherapy with greater side effects (mostly hematologic) is HyperCVAD, often given in the hospital setting, with rituximab and generally to fitter patients (some of which are over 65). HyperCVAD is becoming popular and showing promising results, especially with rituximab. It can be used on some elderly (over 65) patients, but seems only beneficial when the baseline Beta-2-MG blood test was normal. It is showing better complete remissions (CR) and progression-free survival (PFS) than CHOP regimens. A less intensive option is bendamustine with rituximab.[23]
Second line treatment may include fludarabine, combined with cyclophosphamide and/or mitoxantrone, usually with rituximab. Cladribine and clofarabine are two other medications being investigated in MCL. A relatively new regimen that uses old medications is PEP-C, which includes relatively small, daily doses of prednisone, etoposide, procarbazine, and cyclophosphamide, taken orally, has proven effective for relapsed patients. According to Dr. John Leonard, PEP-C may have anti-angiogenetic properties,[24][25] something that he and his colleagues are testing through an ongoing drug trial.[26]
Another approach involves using very high doses of chemotherapy, sometimes combined with total body irradiation (TBI), in an attempt to destroy all evidence of the disease. The downside to this is the destruction of the patient's entire immune system as well, requiring rescue by transplantation of a new immune system (hematopoietic stem cell transplantation), using either autologous stem cell transplantation, or those from a matched donor (an allogeneic stem cell transplant). A presentation at the December 2007 American Society of Hematology (ASH) conference by Christian Geisler, chairman of the Nordic Lymphoma Group[27] claimed that according to trial results, mantle cell lymphoma is potentially curable with very intensive chemo-immunotherapy followed by a stem cell transplant, when treated upon first presentation of the disease.[28][29]
These results seem to be confirmed by a large trial of the European Mantle Cell Lymphoma Network indicating that induction regimens containing monoclonal antibodies and high dose cytarabine followed by autologous stem cell transplantation should become the standard of care of MCL patients up to approximately 65 years of age.[30][31]
A study released in April 2013 showed that patients with previously untreated indolent lymphoma, bendamustine plus rituximab can be considered as a preferred first-line treatment approach to R-CHOP because of increased progression-free survival and fewer toxic effects.[32]
Immunotherapy
Immune-based therapy is dominated by the use of the rituximab monoclonal antibody, sold under the trade name Rituxan (or as Mabthera in Europe and Australia). Rituximab may have good activity against MCL as a single agent, but it is typically given in combination with chemotherapies, which prolongs response duration. There are newer[when?] variations on monoclonal antibodies combined with radioactive molecules known as radioimmunotherapy. These include Zevalin and Bexxar. Rituximab has also been used in small numbers of patients in combination with thalidomide with some effect.[33] In contrast to these antibody-based 'passive' immunotherapies, the field of 'active' immunotherapy tries to activate a patient's immune system to specifically eliminate their own tumor cells. Examples of active immunotherapy include cancer vaccines, adoptive cell transfer, and immunotransplant, which combines vaccination and autologous stem cell transplant. As of 2023, active immunotherapies are not currently a standard of care,[5] but numerous clinical trials are ongoing.[34][35][36]
Targeted therapy
Two Bruton tyrosine kinase inhibitors (BTKi), one In November 2013, ibrutinib (brand name Imbruvica, Pharmacyclics LLC) and one in October 2017, acalabrutinib (brand name Calquence, AstraZeneca Pharmaceuticals LP) were approved in the United States for treating mantle cell lymphoma.[37] However, although these medications are beneficial their duration is short and patients typically relapse.[5]
In November 2019, zanubrutinib (Brukinsa) was approved in the United States with an indication for the treatment of adults with mantle cell lymphoma who have received at least one prior therapy.[38]
Pirtobrutinib (Jaypirca) was approved for medical use in the United States in January 2023.[39]
Gene therapy
Brexucabtagene autoleucel (Tecartus) was approved for medical use in the United States in July 2020, with an indication for the treatment of adults with relapsed or refractory mantle cell lymphoma.[40][41][42] It was approved for medical use in the European Union in December 2020.[43]
Each dose of brexucabtagene autoleucel is a customized treatment created using the recipient's own immune system to help fight the lymphoma.[40] The recipient's T cells, a type of white blood cell, are collected and genetically modified to include a new gene that facilitates the targeting and killing of the lymphoma cells.[40] These modified T cells are then infused back into the recipient.[40]
Prognosis
Recent[when?] clinical advances in mantle cell lymphoma (MCL) have seen standard‐of‐care treatment algorithms transformed. Frontline rituximab combination therapy, high dose cytarabine‐based induction in younger patients and, more recently,[when?] Bruton Tyrosine Kinase (BTK) inhibitors in the relapse setting have all demonstrated survival advantage in clinical trials (Wang et al., 2013; Eskelund et al., 2016; Rule et al., 2016). Over the last[when?] 15 years these practices have gradually become embedded in clinical practice and real‐world data has observed corresponding improvements in patient survival (Abrahamsson et al., 2014; Leux et al., 2014).[44]
The overall 5-year survival rate for MCL is generally 50%[45] (advanced stage MCL) to 70%[46] (for limited-stage MCL).
Prognosis for individuals with MCL is problematic and indexes do not work well because most patients present at the advanced stage disease.[10] Staging is used but is not very informative, since the malignant B-cells can travel freely though the lymphatic system and therefore most patients are at stage III or IV at diagnosis. Prognosis is not strongly affected by staging in MCL and the concept of metastasis does not really apply.[47]
The Mantle Cell Lymphoma International Prognostic Index (MIPI) was derived from a data set of 455 advanced stage MCL patients treated in series of clinical trials in Germany/Europe. Of the evaluable population, approximately 18% were treated with high-dose therapy and stem cell transplantation in first remission. The MIPI is able to classify patients into three risk groups: low risk (median survival not reached after median 32 months follow-up and 5-year OS rate of 60%), intermediate risk (median survival 51 months) and high risk (median survival 29 months). In addition to the 4 independent prognostic factors included in the model, the cell proliferation index (Ki-67) was also shown to have additional prognostic relevance. When the Ki67 is available, a biologic MIPI can be calculated.[48]
MCL is one of the few non-Hodgkin's lymphomas that can cross the boundary into the brain, yet it can be treated in that event.[medical citation needed]
There are a number of prognostic indicators that have been studied. There is not universal agreement on their importance or usefulness in prognosis.[47]
Ki-67 is an indicator of how fast cells mature and is expressed in a range from about 10% to 90%. The lower the percentage, the lower the speed of maturity, and the more indolent the disease. Katzenberger et al. graphs survival versus time for subsets of patients with varying Ki-67 indices. He shows median survival times of about one year for 61–90% Ki-67 and nearly 4 years for 5–20% Ki-67 index.[49]
MCL cell types can aid in prognosis in a subjective way. Blastic is a larger cell type. Diffuse is spread through the node. Nodular are small groups of collected cells spread through the node. Diffuse and nodular are similar in behavior. Blastic is faster growing and it is harder to get long remissions. It has been suggested that in time, some non-blastic MCL transforms to blastic; however, this model has the assumption that increasing genetic alterations lead to the loss of cell cycle control, the higher proliferation rate, and thus to blastoid features. But blastoid features are frequently seen at initial presentation in some patients, whereas other cases remain morphologically stable classical MCL throughout the duration of the disease.[50] Although survival of most blastic patients is shorter, some data shows that 25% of blastic MCL patients survive to 5 years.[50] That is longer than diffuse type and almost as long as nodular (almost 7 yrs).[medical citation needed]
Beta-2 microglobulin is another risk factor in MCL used primarily for transplant patients. Values less than three have yielded 95% overall survival to six years for auto SCT where over three yields a median of 44 most overall survival for auto SCT (Khouri 03). This is not yet[when?] fully validated.[medical citation needed]
Testing for high levels of lactate dehydrogenase in patients with non-Hodgkin's lymphoma is useful because it is released when body tissues break down for any reason. While it cannot be used as a sole means of diagnosing non-Hodgkin's lymphoma, it is a marker for tracking tumor burden in those diagnosed by other means. The normal range is approximately between 140 and 280 U/L [51] but the clinical interpretation will depend upon the patient's symptoms.
↑ Rummel MJ, Niederle N, Maschmeyer G, etal. (April 2013). "Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial". Lancet. 381 (9873): 1203–10. doi:10.1016/S0140-6736(12)61763-2. PMID23433739. S2CID27886488.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.