
Polydactyly or polydactylism, also known as hyperdactyly, is an anomaly in humans and animals resulting in supernumerary fingers and/or toes. Polydactyly is the opposite of oligodactyly.

Aniridia is the absence of the iris, a muscular structure that opens and closes the pupil to allow light into the eye. It is also responsible for eye color. Without it, the central eye appears all black. It can be congenital, in which both eyes are usually involved, or caused by a penetrant injury. Isolated aniridia is a congenital disorder which is not limited to a defect in iris development, but is a panocular condition with macular and optic nerve hypoplasia, cataract, and corneal changes. Vision may be severely compromised and the disorder is frequently associated with a number of ocular complications: nystagmus, amblyopia, buphthalmos, and cataract. Aniridia in some individuals occurs as part of a syndrome, such as WAGR syndrome, or Gillespie syndrome.

Fraser syndrome is an autosomal recessive congenital disorder, identified by several developmental anomalies. Fraser syndrome is named for the geneticist George R. Fraser, who first described the syndrome in 1962.

Nevoid basal-cell carcinoma syndrome (NBCCS) is an inherited medical condition involving defects within multiple body systems such as the skin, nervous system, eyes, endocrine system, and bones. People with this syndrome are particularly prone to developing a common and usually non-life-threatening form of non-melanoma skin cancer. About 10% of people with the condition do not develop basal-cell carcinomas (BCCs).

Duane-radial ray syndrome, also known as Okihiro Syndrome, is a rare autosomal dominant disorder that primarily affects the eyes and causes abnormalities of bones in the arms and hands. This disorder is considered to be a SALL4-related disorder due to the SALL4 gene mutations leading to these abnormalities. It is diagnosed by clinical findings on a physical exam as well as genetic testing and imaging. After being diagnosed, there are other evaluations that one may go through in order to determine the extent of the disease. There are various treatments for the symptoms of this disorder.

Young–Simpson syndrome (YSS) is a rare congenital disorder with symptoms including hypothyroidism, heart defects, facial dysmorphism, cryptorchidism in males, hypotonia, intellectual disability, and postnatal growth retardation.
1q21.1 duplication syndrome or 1q21.1 (recurrent) microduplication is a rare aberration of chromosome 1.

Distal 18q- is a genetic condition caused by a deletion of genetic material within one of the two copies of chromosome 18. The deletion involves the distal section of 18q and typically extends to the tip of the long arm of chromosome 18.

Ectrodactyly, split hand, or cleft hand involves the deficiency or absence of one or more central digits of the hand or foot and is also known as split hand/split foot malformation (SHFM). The hands and feet of people with ectrodactyly (ectrodactyls) are often described as "claw-like" and may include only the thumb and one finger with similar abnormalities of the feet.
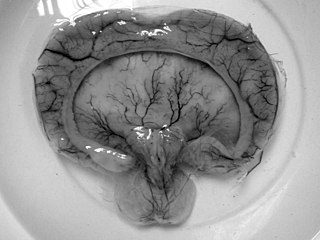
Young–Madders syndrome, alternatively known as Pseudotrisomy 13 syndrome or holoprosencephaly–polydactyly syndrome, is a genetic disorder resulting from defective and duplicated chromosomes which result in holoprosencephaly, polydactyly, facial malformations and intellectual disability, with a significant variance in the severity of symptoms being seen across known cases. Many cases often suffer with several other genetic disorders, and some have presented with hypoplasia, cleft lip, cardiac lesions and other heart defects. In one case in 1991 and another in 2000 the condition was found in siblings who were the product of incest. Many cases are diagnosed prenatally and often in siblings. Cases are almost fatal in the prenatal stage with babies being stillborn.
13q deletion syndrome is a rare genetic disease caused by the deletion of some or all of the large arm of human chromosome 13. Depending upon the size and location of the deletion on chromosome 13, the physical and mental manifestations will vary. It has the potential to cause intellectual disability and congenital malformations that affect a variety of organ systems. Because of the rarity of the disease in addition to the variations in the disease, the specific genes that cause this disease are unknown. This disease is also known as:
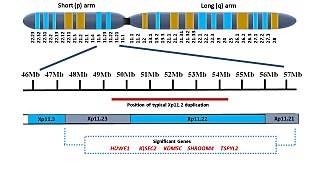
Xp11.2 duplication is a genomic variation marked by the duplication of an X chromosome region on the short arm p at position 11.2, defined by standard karyotyping (G-banding). This gene-rich, rearrangement prone region can be further divided into three loci - Xp11.21, Xp11.22 and Xp11.23. The duplication could involve any combination of these three loci. While the length of the duplication can vary from 0.5Mb to 55 Mb, most duplications measure about 4.5Mb and typically occur in the region of 11.22-11.23. Most affected females show preferential activation of the duplicated X chromosome. Features of affected individuals vary significantly, even among members of the same family. The Xp11.2 duplication can be 'silent' - presenting no obvious symptoms in carriers - which is known from the asymptomatic parents of affected children carrying the duplication. The common symptoms include intellectual disabilities, speech delay and learning difficulties, while in rare cases, children have seizures and a recognizable brain wave pattern when assessed by EEG (electroencephalography).

17q12 microdeletion syndrome, also known as 17q12 deletion syndrome, is a rare chromosomal anomaly caused by the deletion of a small amount of material from a region in the long arm of chromosome 17. It is typified by deletion of the HNF1B gene, resulting in kidney abnormalities and renal cysts and diabetes syndrome. It also has neurocognitive effects, and has been implicated as a genetic factor for autism and schizophrenia.
Filippi syndrome, also known as Syndactyly Type I with Microcephaly and Mental Retardation, is a very rare autosomal recessive genetic disease. Only a very limited number of cases have been reported to date. Filippi Syndrome is associated with diverse symptoms of varying severity across affected individuals, for example malformation of digits, craniofacial abnormalities, intellectual disability, and growth retardation. The diagnosis of Filippi Syndrome can be done through clinical observation, radiography, and genetic testing. Filippi Syndrome cannot be cured directly as of 2022, hence the main focus of treatments is on tackling the symptoms observed on affected individuals. It was first reported in 1985.

Familial opposable triphalangeal thumb duplication is a limb malformation syndrome and a type of pre-axial polydactyly, characterized by having duplicated opposable triphalangeal thumbs. This condition can be a symptom of other genetic disorders, such as Holt–Oram syndrome and Fanconi anemia. This trait is autosomal dominant and often runs in families. Sometimes big toe duplication, post-axial polydactyly, and syndactyly of the hand and feet can occur alongside this malformation Approximately 20 families with the condition have been described in medical literature.

Waardenburg anophthalmia syndrome is a rare autosomal recessive genetic disorder which is characterized by either microphthalmia or anophthalmia, osseous synostosis, ectrodactylism, polydactylism, and syndactylism. So far, 29 cases from families in Brazil, Italy, Turkey, and Lebanon have been reported worldwide. This condition is caused by homozygous mutations in the SMOC1 gene, in chromosome 14.

Metacarpal synostosis is a rare congenital difference which is characterized by the fusion of 2 metacarpals of the hand, which are usually shortened. It is most commonly seen as a fusion of the 4th and 5th metacarpals. It is a type of non-syndromic syndactyly/synostosis. Autosomal dominant and X-linked recessive inheritance patterns have been reported.
Gollop-Wolfgang complex is a very rare genetic disorder which is characterized by skeletal and digital anomalies.

Heart-hand syndrome, Slovenian type is a rare autosomal dominant genetic disorder belonging to the heart-hand syndromes.
