Related Research Articles

A chromosome is a long DNA molecule with part or all of the genetic material of an organism. In most chromosomes the very long thin DNA fibers are coated with packaging proteins; in eukaryotic cells the most important of these proteins are the histones. These proteins, aided by chaperone proteins, bind to and condense the DNA molecule to maintain its integrity. These chromosomes display a complex three-dimensional structure, which plays a significant role in transcriptional regulation.

Turner syndrome (TS), also known as 45,X, or 45,X0, is a genetic disorder caused by a sex chromosome monosomy, compared to the two sex chromosomes in most people. Signs and symptoms vary among those affected. Often, a short and webbed neck, low-set ears, low hairline at the back of the neck, short stature, and swollen hands and feet are seen at birth. Typically, those affected do not develop menstrual periods or breasts without hormone treatment and are unable to have children without reproductive technology. Heart defects, diabetes, and low thyroid hormone occur in the disorder more frequently than average. Most people with Turner syndrome have normal intelligence; however, many have problems with spatial visualization that may be needed in order to learn mathematics. Vision and hearing problems also occur more often than average.

In genetics, a deletion is a mutation in which a part of a chromosome or a sequence of DNA is left out during DNA replication. Any number of nucleotides can be deleted, from a single base to an entire piece of chromosome. Some chromosomes have fragile spots where breaks occur, which result in the deletion of a part of the chromosome. The breaks can be induced by heat, viruses, radiation, or chemical reactions. When a chromosome breaks, if a part of it is deleted or lost, the missing piece of chromosome is referred to as a deletion or a deficiency.

A small supernumerary marker chromosome (sSMC) is an abnormal extra chromosome. It contains copies of parts of one or more normal chromosomes and like normal chromosomes is located in the cell's nucleus, is replicated and distributed into each daughter cell during cell division, and typically has genes which may be expressed. However, it may also be active in causing birth defects and neoplasms. The sSMC's small size makes it virtually undetectable using classical cytogenetic methods: the far larger DNA and gene content of the cell's normal chromosomes obscures those of the sSMC. Newer molecular techniques such as fluorescence in situ hybridization, next generation sequencing, comparative genomic hybridization, and highly specialized cytogenetic G banding analyses are required to study it. Using these methods, the DNA sequences and genes in sSMCs are identified and help define as well as explain any effect(s) it may have on individuals.
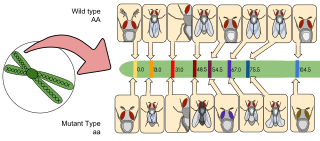
Gene mapping or genome mapping describes the methods used to identify the location of a gene on a chromosome and the distances between genes. Gene mapping can also describe the distances between different sites within a gene.
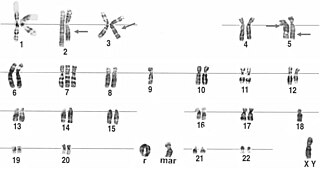
A ring chromosome is an aberrant chromosome whose ends have fused together to form a ring. Ring chromosomes were first discovered by Lilian Vaughan Morgan in 1926. A ring chromosome is denoted by the symbol r in human genetics and R in Drosophila genetics. Ring chromosomes may form in cells following genetic damage by mutagens like radiation, but they may also arise spontaneously during development.

Medical genetics is the branch of medicine that involves the diagnosis and management of hereditary disorders. Medical genetics differs from human genetics in that human genetics is a field of scientific research that may or may not apply to medicine, while medical genetics refers to the application of genetics to medical care. For example, research on the causes and inheritance of genetic disorders would be considered within both human genetics and medical genetics, while the diagnosis, management, and counselling people with genetic disorders would be considered part of medical genetics.
The Pallister–Killian syndrome (PKS), also termed tetrasomy 12p mosaicism or the Pallister mosaic aneuploidy syndrome, is an extremely rare and severe genetic disorder. PKS is due to the presence of an extra and abnormal chromosome termed a small supernumerary marker chromosome (sSMC). sSMCs contain copies of genetic material from parts of virtually any other chromosome and, depending on the genetic material they carry, can cause various genetic disorders and neoplasms. The sSMC in PKS consists of multiple copies of the short arm of chromosome 12. Consequently, the multiple copies of the genetic material in the sSMC plus the two copies of this genetic material in the two normal chromosome 12's are overexpressed and thereby cause the syndrome. Due to a form of genetic mosaicism, however, individuals with PKS differ in the tissue distributions of their sSMC and therefore show different syndrome-related birth defects and disease severities. For example, individuals with the sSMC in their heart tissue are likely to have cardiac structural abnormalities while those without this sSMC localization have a structurally normal heart.
A dicentric chromosome is an abnormal chromosome with two centromeres. It is formed through the fusion of two chromosome segments, each with a centromere, resulting in the loss of acentric fragments and the formation of dicentric fragments. The formation of dicentric chromosomes has been attributed to genetic processes, such as Robertsonian translocation and paracentric inversion. Dicentric chromosomes have important roles in the mitotic stability of chromosomes and the formation of pseudodicentric chromosomes. Their existence has been linked to certain natural phenomena such as irradiation and have been documented to underlie certain clinical syndromes, notably Kabuki syndrome. The formation of dicentric chromosomes and their implications on centromere function are studied in certain clinical cytogenetics laboratories.

Isodicentric 15, also called marker chromosome 15 syndrome, idic(15), partial tetrasomy 15q, or inverted duplication 15, is a chromosome abnormality in which a child is born with extra genetic material from chromosome 15. People with idic(15) are typically born with 47 chromosomes in their body cells, instead of the normal 46. The extra chromosome, which is classified as a small supernumerary marker chromosome, is made up of a piece of chromosome 15 that has been duplicated end-to-end like a mirror image. It is the presence of this extra genetic material that is thought to account for the symptoms seen in some people with idic(15). Individuals with idic(15) have a total of four copies of this chromosome 15 region instead of the usual two copies. The term isodicentric refers to a duplication and inversion of a centromere-containing chromosomal segment.
Emanuel syndrome, also known as derivative 22 syndrome, or der(22) syndrome, is a rare disorder associated with multiple congenital anomalies, including profound intellectual disability, preauricular skin tags or pits, and conotruncal heart defects. It can occur in offspring of carriers of the constitutional chromosomal translocation t(11;22)(q23;q11), owing to a 3:1 meiotic malsegregation event resulting in partial trisomy of chromosomes 11 and 22. An unbalanced translocation between chromosomes 11 and 22 is described as Emanuel syndrome. It was first described in 1980 by American medical researchers Beverly S. Emanuel and Elaine H. Zackai, and a consortium of European scientists the same year.

Tetrasomy 18p is a genetic condition that is caused by the presence of an isochromosome composed of two copies of the short arm of chromosome 18 in addition to the two normal copies of the chromosome. It is characterized by multiple medical and developmental concerns.

Cat eye syndrome (CES) or Schmid–Fraccaro syndrome is a rare condition caused by an abnormal extra chromosome, i.e. a small supernumerary marker chromosome. This chromosome consists of the entire short arm and a small section of the long arm of chromosome 22. In consequence, individuals with the cat eye syndrome have three (trisomic) or four (tetrasomic) copies of the genetic material contained in the abnormal chromosome instead of the normal two copies. The prognosis for patients with CES varies depending on the severity of the condition and their associated signs and symptoms, especially when heart or kidney abnormalities are seen.

Structural maintenance of chromosomes protein 1A (SMC1A) is a protein that in humans is encoded by the SMC1A gene. SMC1A is a subunit of the cohesin complex which mediates sister chromatid cohesion, homologous recombination and DNA looping. In somatic cells, cohesin is formed of SMC1A, SMC3, RAD21 and either SA1 or SA2 whereas in meiosis, cohesin is formed of SMC3, SMC1B, REC8 and SA3.
X-linked intellectual disability refers to medical disorders associated with X-linked recessive inheritance that result in intellectual disability.
Tetrasomy 9p is a rare chromosomal disorder characterized by the presence of two extra copies of the short arm of chromosome 9, in addition to the usual two. Symptoms of tetrasomy 9p vary widely among affected individuals but typically include varying degrees of delayed growth, abnormal facial features and intellectual disability. Symptoms of the disorder are comparable to those of trisomy 9p.
9q34 deletion syndrome is a rare genetic disorder. Terminal deletions of chromosome 9q34 have been associated with childhood hypotonia, a distinctive facial appearance and developmental disability. The facial features typically described include arched eyebrows, small head circumference, midface hypoplasia, prominent jaw and a pouting lower lip. Individuals with this disease may often have speech impediments, such as speech delays. Other characteristics of this disease include: epilepsy, congenital and urogenital defects, microcephaly, corpulence, and psychiatric disorders. From analysis of chromosomal breakpoints, as well as gene sequencing in suggestive cases, Kleefstra and colleagues identified EHMT1 as the causative gene. This gene is responsible for producing the protein histone methyltransferase which functions to alter histones. Ultimately, histone methyltransferases are important in deactivating certain genes, needed for proper growth and development. Moreover, a frameshift, missense, or nonsense error in the coding sequence of EHMT1 can result in this condition in an individual.
About 10–15% of human couples are infertile, unable to conceive. In approximately in half of these cases, the underlying cause is related to the male. The underlying causative factors in the male infertility can be attributed to environmental toxins, systemic disorders such as, hypothalamic–pituitary disease, testicular cancers and germ-cell aplasia. Genetic factors including aneuploidies and single-gene mutations are also contributed to the male infertility. Patients with nonobstructive azoospermia or oligozoospermia show microdeletions in the long arm of the Y chromosome and/or chromosomal abnormalities, each with the respective frequency of 9.7% and 13%. A large percentage of human male infertility is estimated to be caused by mutations in genes involved in primary or secondary spermatogenesis and sperm quality and function. Single-gene defects are the focus of most research carried out in this field.
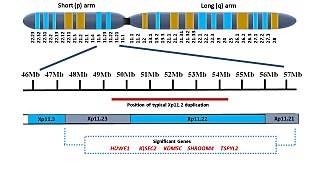
Xp11.2 duplication is a genomic variation marked by the duplication of an X chromosome region on the short arm p at position 11.2, defined by standard karyotyping (G-banding). This gene-rich, rearrangement prone region can be further divided into three loci - Xp11.21, Xp11.22 and Xp11.23. The duplication could involve any combination of these three loci. While the length of the duplication can vary from 0.5Mb to 55 Mb, most duplications measure about 4.5Mb and typically occur in the region of 11.22-11.23. Most affected females show preferential activation of the duplicated X chromosome. Features of affected individuals vary significantly, even among members of the same family. The Xp11.2 duplication can be 'silent' - presenting no obvious symptoms in carriers - which is known from the asymptomatic parents of affected children carrying the duplication. The common symptoms include intellectual disabilities, speech delay and learning difficulties, while in rare cases, children have seizures and a recognizable brain wave pattern when assessed by EEG (electroencephalography).

17q12 microdeletion syndrome, also known as 17q12 deletion syndrome, is a rare chromosomal anomaly caused by the deletion of a small amount of material from a region in the long arm of chromosome 17. It is typified by deletion of the HNF1B gene, resulting in kidney abnormalities and renal cysts and diabetes syndrome. It also has neurocognitive effects, and has been implicated as a genetic factor for autism and schizophrenia.
References
- 1 2 Thompson & Thompson Genetics in Medicine, Chapter 5, 57-74 https://www.clinicalkey.com/#!/content/book/3-s2.0-B9781437706963000054?scrollTo=%23hl0000654
- 1 2 Nelson Textbook of Pediatrics, Chapter 81, 604-627 https://www.clinicalkey.com/#!/content/book/3-s2.0-B9781455775668000818?scrollTo=%23hl0003126
- ↑ Sun M, Zhang H, Li G, Guy CJ, Wang X, Lu X, Gong F, Lee J, Hassed S, Li S (September 2017). "Molecular characterization of 20 small supernumerary marker chromosome cases using array comparative genomic hybridization and fluorescence in situ hybridization". Scientific Reports. 7 (1): 10395. Bibcode:2017NatSR...710395S. doi:10.1038/s41598-017-10466-z. PMC 5583289 . PMID 28871159.
- ↑ Jafari-Ghahfarokhi H, Moradi-Chaleshtori M, Liehr T, Hashemzadeh-Chaleshtori M, Teimori H, Ghasemi-Dehkordi P (2015). "Small supernumerary marker chromosomes and their correlation with specific syndromes". Advanced Biomedical Research. 4: 140. doi: 10.4103/2277-9175.161542 . PMC 4544121 . PMID 26322288.
- ↑ Baldwin, Erin L.; May, Lorraine F.; Justice, April N.; Martin, Christa L.; Ledbetter, David H. (8 February 2008). "Mechanisms and Consequences of Small Supernumerary Marker Chromosomes: From Barbara McClintock to Modern Genetic-Counseling Issues". American Journal of Human Genetics. 82 (2): 398–410. doi:10.1016/j.ajhg.2007.10.013. PMC 2427313 . PMID 18252220.
- https://web.archive.org/web/20060926021351/http://www.chromodisorder.org/sytrix/card_list.php3?dbid=63&id=365 An International System for Human Cytogenetic Nomenclature, Shaffer, L.G., Tommerup N. (eds); S. Karger, Basel 2005