Related Research Articles

Lymphoma is a group of blood and lymph tumors that develop from lymphocytes. The name typically refers to just the cancerous versions rather than all such tumours. Signs and symptoms may include enlarged lymph nodes, fever, drenching sweats, unintended weight loss, itching, and constantly feeling tired. The enlarged lymph nodes are usually painless. The sweats are most common at night.

Anaplastic large-cell lymphoma (ALCL) refers to a group of non-Hodgkin lymphomas in which aberrant T cells proliferate uncontrollably. Considered as a single entity, ALCL is the most common type of peripheral lymphoma and represents ~10% of all peripheral lymphomas in children. The incidence of ALCL is estimated to be 0.25 cases per 100,000 people in the United States of America. There are four distinct types of anaplastic large-cell lymphomas that on microscopic examination share certain key histopathological features and tumor marker proteins. However, the four types have very different clinical presentations, gene abnormalities, prognoses, and/or treatments.
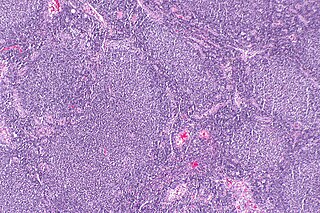
Follicular lymphoma (FL) is a cancer that involves certain types of white blood cells known as lymphocytes. The cancer originates from the uncontrolled division of specific types of B-cells known as centrocytes and centroblasts. These cells normally occupy the follicles in the germinal centers of lymphoid tissues such as lymph nodes. The cancerous cells in FL typically form follicular or follicle-like structures in the tissues they invade. These structures are usually the dominant histological feature of this cancer.
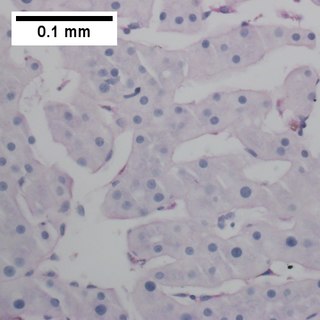
Primary effusion lymphoma (PEL) is classified as a diffuse large B cell lymphoma. It is a rare malignancy of plasmablastic cells that occurs in individuals that are infected with the Kaposi's sarcoma-associated herpesvirus. Plasmablasts are immature plasma cells, i.e. lymphocytes of the B-cell type that have differentiated into plasmablasts but because of their malignant nature do not differentiate into mature plasma cells but rather proliferate excessively and thereby cause life-threatening disease. In PEL, the proliferating plasmablastoid cells commonly accumulate within body cavities to produce effusions, primarily in the pleural, pericardial, or peritoneal cavities, without forming a contiguous tumor mass. In rare cases of these cavitary forms of PEL, the effusions develop in joints, the epidural space surrounding the brain and spinal cord, and underneath the capsule which forms around breast implants. Less frequently, individuals present with extracavitary primary effusion lymphomas, i.e., solid tumor masses not accompanied by effusions. The extracavitary tumors may develop in lymph nodes, bone, bone marrow, the gastrointestinal tract, skin, spleen, liver, lungs, central nervous system, testes, paranasal sinuses, muscle, and, rarely, inside the vasculature and sinuses of lymph nodes. As their disease progresses, however, individuals with the classical effusion-form of PEL may develop extracavitary tumors and individuals with extracavitary PEL may develop cavitary effusions.

Intravascular lymphomas (IVL) are rare cancers in which malignant lymphocytes proliferate and accumulate within blood vessels. Almost all other types of lymphoma involve the proliferation and accumulation of malignant lymphocytes in lymph nodes, other parts of the lymphatic system, and various non-lymphatic organs but not in blood vessels.
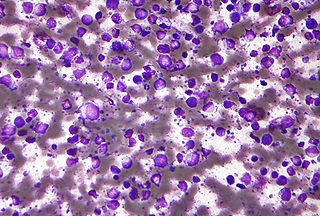
Diffuse large B-cell lymphoma (DLBCL) is a cancer of B cells, a type of lymphocyte that is responsible for producing antibodies. It is the most common form of non-Hodgkin lymphoma among adults, with an annual incidence of 7–8 cases per 100,000 people per year in the US and UK. This cancer occurs primarily in older individuals, with a median age of diagnosis at ~70 years, although it can occur in young adults and, in rare cases, children. DLBCL can arise in virtually any part of the body and, depending on various factors, is often a very aggressive malignancy. The first sign of this illness is typically the observation of a rapidly growing mass or tissue infiltration that is sometimes associated with systemic B symptoms, e.g. fever, weight loss, and night sweats.

Marginal zone lymphomas, also known as marginal zone B-cell lymphomas (MZLs), are a heterogeneous group of lymphomas that derive from the malignant transformation of marginal zone B-cells. Marginal zone B cells are innate lymphoid cells that normally function by rapidly mounting IgM antibody immune responses to antigens such as those presented by infectious agents and damaged tissues. They are lymphocytes of the B-cell line that originate and mature in secondary lymphoid follicles and then move to the marginal zones of mucosa-associated lymphoid tissue (MALT), the spleen, or lymph nodes. Mucosa-associated lymphoid tissue is a diffuse system of small concentrations of lymphoid tissue found in various submucosal membrane sites of the body such as the gastrointestinal tract, mouth, nasal cavity, pharynx, thyroid gland, breast, lung, salivary glands, eye, skin and the human spleen.

Follicular dendritic cell sarcoma (FDCS) is an extremely rare neoplasm. While the existence of FDC tumors was predicted by Lennert in 1978, the tumor wasn't fully recognized as its own cancer until 1986 after characterization by Monda et al. It accounts for only 0.4% of soft tissue sarcomas, but has significant recurrent and metastatic potential and is considered an intermediate grade malignancy. The major hurdle in treating FDCS has been misdiagnosis. It is a newly characterized cancer, and because of its similarities in presentation and markers to lymphoma, both Hodgkin and Non-Hodgkin subtypes, diagnosis of FDCS can be difficult. With recent advancements in cancer biology better diagnostic assays and chemotherapeutic agents have been made to more accurately diagnose and treat FDCS.

Extranodal NK/T-cell lymphoma, nasal type (ENKTCL-NT) is a rare type of lymphoma that commonly involves midline areas of the nasal cavity, oral cavity, and/or pharynx At these sites, the disease often takes the form of massive, necrotic, and extremely disfiguring lesions. However, ENKTCL-NT can also involve the eye, larynx, lung, gastrointestinal tract, skin, and various other tissues. ENKTCL-NT mainly affects adults; it is relatively common in Asia and to lesser extents Mexico, Central America, and South America but is rare in Europe and North America. In Korea, ENKTCL-NT often involves the skin and is reported to be the most common form of cutaneous lymphoma after mycosis fungoides.

Peripheral T-cell lymphoma not otherwise specified (PTCL-NOS), is a subtype of peripheral T-cell lymphoma. Peripheral T-cell lymphoma (PTCL) is defined as a diverse group of aggressive lymphomas that develop from mature-stage white blood cells called T-cells and natural killer cells. PTCL is a type of non-Hodgkin's lymphoma (NHL). PTCL specifically affects T-cells rather than B-cells, and results when T-cells develop and grow abnormally.

Plasmablastic lymphoma (PBL) is a type of large B-cell lymphoma recognized by the World Health Organization (WHO) in 2017 as belonging to a subgroup of lymphomas termed lymphoid neoplasms with plasmablastic differentiation. The other lymphoid neoplasms within this subgroup are: plasmablastic plasma cell lymphoma ; primary effusion lymphoma that is Kaposi's sarcoma-associated herpesvirus positive or Kaposi's sarcoma-associated Herpesvirus negative; anaplastic lymphoma kinase-positive large B-cell lymphoma; and human herpesvirus 8-positive diffuse large B-cell lymphoma, not otherwise specified. All of these lymphomas are malignancies of plasmablasts, i.e. B-cells that have differentiated into plasmablasts but because of their malignant nature: fail to differentiate further into mature plasma cells; proliferate excessively; and accumulate in and injure various tissues and organs.
Epstein–Barr virus–associated lymphoproliferative diseases are a group of disorders in which one or more types of lymphoid cells, i.e. B cells, T cells, NK cells, and histiocytic-dendritic cells, are infected with the Epstein–Barr virus (EBV). This causes the infected cells to divide excessively, and is associated with the development of various non-cancerous, pre-cancerous, and cancerous lymphoproliferative disorders (LPDs). These LPDs include the well-known disorder occurring during the initial infection with the EBV, infectious mononucleosis, and the large number of subsequent disorders that may occur thereafter. The virus is usually involved in the development and/or progression of these LPDs although in some cases it may be an "innocent" bystander, i.e. present in, but not contributing to, the disease.
Indolent T cell lymphoproliferative disorder of the gastrointestinal tract or Indolent T cell lymphoproliferative disorder of the GI tract (ITCLD-GT) is a rare and recently recognized disorder in which mature T cell lymphocytes accumulation abnormally in the gastrointestinal tract. This accumulation causes various lesions in the mucosal layer lining the GI tract. Individuals with ITCLD-GT commonly complain of chronic GI tract symptoms such as nausea, vomiting, diarrhea, abdominal pain, and rectal bleeding.
Monomorphic epitheliotropic intestinal T cell lymphoma (MEITL) is an extremely rare peripheral T-cell lymphoma that involves the malignant proliferation of a type of lymphocyte, the T cell, in the gastrointestinal tract. Over time, these T cells commonly spread throughout the mucosal lining of a portion of the GI tract, lead to GI tract nodules and ulcerations, and cause symptoms such as abdominal pain, weight loss, diarrhea, obstruction, bleeding, and/or perforation.
In situ lymphoid neoplasia is a precancerous condition newly classified by the World Health Organization in 2016. The Organization recognized two subtypes of ISLN: in situ follicular neoplasia (ISFN) and in situ mantle cell neoplasia (ISMCL). ISFN and ISMCL are pathological accumulations of lymphocytes in the germinal centers and mantle zones, respectively, of the follicles that populate lymphoid organs such as lymph nodes. These lymphocytes are monoclonal B-cells that may develop into follicular (FL) and mantle cell (MCL) lymphomas, respectively.
Pediatric-type follicular lymphoma (PTFL) is a disease in which malignant B-cells accumulate in, overcrowd, and cause the expansion of the lymphoid follicles in, and thereby enlargement of the lymph nodes in the head and neck regions and, less commonly, groin and armpit regions. The disease accounts for 1.5% to 2% of all the lymphomas that occur in the pediatric age group.
Duodenal-type follicular lymphoma (DFL) is a form of lymphoma in which certain lymphocyte types, the B-cell-derived centrocytes and centroblasts, form lymph node follicle-like structures principally in the duodenum and other parts of the small intestine. It is an indolent disease which on rare occasions progresses to a more aggressive lymphoma that spreads beyond these originally involved sites.
T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL) is a malignancy of B cells. B-cells are lymphocytes that normally function in the humoral immunity component of the adaptive immune system by secreting antibodies that, for example, bind to and neutralize invasive pathogens. Among the various forms of B-cell lymphomas, THRLBCL is a rarely occurring subtype of the diffuse large B-cell lymphomas (DLBCL). DLBCL are a large group of lymphomas that account for ~25% of all non-Hodgkin lymphomas worldwide. THRLBCL is distinguished from the other DLBCL subtypes by the predominance of non-malignant T-cell lymphocytes and histiocytes over malignant B-cells in its tumors and tissue infiltrates.
Primary cutaneous diffuse large B-cell lymphoma, leg type (PCDLBCL-LT) is a cutaneous lymphoma skin disease that occurs mostly in elderly females. In this disease, B cells become malignant, accumulate in the dermis and subcutaneous tissue below the dermis to form red and violaceous skin nodules and tumors. These lesions typically occur on the lower extremities but in uncommon cases may develop on the skin at virtually any other site. In ~10% of cases, the disease presents with one or more skin lesions none of which are on the lower extremities; the disease in these cases is sometimes regarded as a variant of PCDLBL, LT termed primary cutaneous diffuse large B-cell lymphoma, other (PCDLBC-O). PCDLBCL, LT is a subtype of the diffuse large B-cell lymphomas (DLBCL) and has been thought of as a cutaneous counterpart to them. Like most variants and subtypes of the DLBCL, PCDLBCL, LT is an aggressive malignancy. It has a 5-year overall survival rate of 40–55%, although the PCDLBCL-O variant has a better prognosis than cases in which the legs are involved.
Mature T-cell lymphoma, also called peripheral T-cell lymphoma, is a group of rare, aggressive lymphomas that develop from mature white blood cells and originate from lymphoid tissues outside of the bone marrow. Mature T-cell lymphoma is under the category of non-Hodgkin lymphoma. Mature T-cell lymphomas account for 10% to 15% of all lymphomas and is more common in Asia than in Europe and America. Its common subtypes include angioimmunoblastic T-cell lymphoma, anaplastic large cell lymphoma and peripheral T-cell lymphoma not otherwise specified. While different subtypes have variable symptoms, common symptoms include enlarged painless lymph nodes, fever, weight loss, rash and night sweats.
References
- 1 2 Summers TA, Rush W, Aguilera N, Lupton G (October 2009). "Cutaneous involvement in the lymphoepithelioid variant of peripheral T-cell lymphoma, unspecified (Lennert lymphoma). Report of a case and review of the literature". Journal of Cutaneous Pathology. 36 Suppl 1: 25–30. doi:10.1111/j.1600-0560.2008.01203.x. PMID 19775391.
- 1 2 3 Hartmann S, Agostinelli C, Klapper W, Korkolopoulou P, Koch K, Marafioti T, Piccaluga PP, Patsouris E, Pileri S, Hansmann ML (December 2011). "Revising the historical collection of epithelioid cell-rich lymphomas of the Kiel Lymph Node Registry: what is Lennert's lymphoma nowadays?". Histopathology. 59 (6): 1173–82. doi:10.1111/j.1365-2559.2011.04069.x. PMID 22175897.
- 1 2 3 4 5 6 Feller AC, Griesser GH, Mak TW, Lennert K (September 1986). "Lymphoepithelioid lymphoma (Lennert's lymphoma) is a monoclonal proliferation of helper/inducer T cells". Blood. 68 (3): 663–7. doi:10.1182/blood.V68.3.663.663. PMID 2943330.
- ↑ Lennert K: Zur Histologischen Diagnose der Lymphogranulomatose. Frankfurt, FRG, Habil-Schrift, 1952
- 1 2 3 4 5 6 Yin Y, Liu H, Luo M, Yu G, Yin W, Li P (February 2023). "Primary extranodal soft tissue Lennert lymphoma (lymphoepithelioid variant of peripheral T-cell lymphoma, unspecified): a case report and review of the literature". Diagnostic Pathology. 18 (1): 12. doi: 10.1186/s13000-023-01297-w . PMC 9896693 . PMID 36737805.
- 1 2 3 Savage KJ, Ferreri AJ, Zinzani PL, Pileri SA (September 2011). "Peripheral T-cell lymphoma--not otherwise specified". Critical Reviews in Oncology/Hematology. 79 (3): 321–9. doi:10.1016/j.critrevonc.2010.07.007. PMID 20702104.
- 1 2 3 4 5 6 7 8 9 10 11 Etebari M, Navari M, Agostinelli C, Visani A, Peron C, Iqbal J, Inghirami G, Piccaluga PP (2019). "Transcriptional Analysis of Lennert Lymphoma Reveals a Unique Profile and Identifies Novel Therapeutic Targets". Frontiers in Genetics. 10: 780. doi: 10.3389/fgene.2019.00780 . PMC 6748072 . PMID 31552092.
- ↑ Kitamura A, Yamashita Y, Sato Y, Hasegawa Y, Kojima H, Nagasawa T, Mori N (October 2005). "Aggressive Lennert's lymphoma: report of three cases in comparison to non-aggressive Lennert's lymphoma". Pathology International. 55 (10): 626–31. doi:10.1111/j.1440-1827.2005.01880.x. PMID 16185292.
- 1 2 Ondrejka SL, Amador C, Climent F, Ng SB, Soma L, Zamo A, Dirnhofer S, Quintanilla-Martinez L, Wotherspoon A, Leoncini L, de Leval L (September 2023). "Follicular helper T-cell lymphomas: disease spectrum, relationship with clonal hematopoiesis, and mimics. A report of the 2022 EA4HP/SH lymphoma workshop". Virchows Archiv. 483 (3): 349–365. doi:10.1007/s00428-023-03607-5. PMC 10541838 . PMID 37500795.
- 1 2 3 4 5 6 7 8 9 10 Kurita D, Miyoshi H, Yoshida N, Sasaki Y, Kato S, Niino D, Sugita Y, Hatta Y, Takei M, Makishima M, Ohshima K (September 2016). "A Clinicopathologic Study of Lennert Lymphoma and Possible Prognostic Factors: The Importance of Follicular Helper T-cell Markers and the Association With Angioimmunoblastic T-cell Lymphoma". The American Journal of Surgical Pathology. 40 (9): 1249–60. doi:10.1097/PAS.0000000000000694. PMID 27428734.
- 1 2 3 4 5 6 Weisenburger DD, Savage KJ, Harris NL, Gascoyne RD, Jaffe ES, MacLennan KA, Rüdiger T, Pileri S, Nakamura S, Nathwani B, Campo E, Berger F, Coiffier B, Kim WS, Holte H, Federico M, Au WY, Tobinai K, Armitage JO, Vose JM (March 2011). "Peripheral T-cell lymphoma, not otherwise specified: a report of 340 cases from the International Peripheral T-cell Lymphoma Project". Blood. 117 (12): 3402–8. doi:10.1182/blood-2010-09-310342. hdl: 11380/662446 . PMID 21270441.
- ↑ Kim JH, Lee SK, Kim HY, Uh JA, Lee JH, Kim MS, Lee UH (February 2022). "Rare Case of Double-Positive CD4/CD8 Immunophenotype in Lennert Lymphoma With Cutaneous Involvement: A Case Report". The American Journal of Dermatopathology. 44 (2): 121–125. doi:10.1097/DAD.0000000000002011. PMID 34816803.
- 1 2 Zhan HQ, Li XQ, Zhu XZ, Lu HF, Zhou XY, Chen Y (April 2011). "Expression of follicular helper T cell markers in nodal peripheral T cell lymphomas: a tissue microarray analysis of 162 cases". Journal of Clinical Pathology. 64 (4): 319–24. doi:10.1136/jcp.2010.084459. PMID 21330314.
- 1 2 Seighali N, Shafiee A, Rafiee MA, Aminzade D, Mozhgani SH (May 2023). "Human T-cell lymphotropic virus type 1 (HTLV-1) proposed vaccines: a systematic review of preclinical and clinical studies". BMC Infectious Diseases. 23 (1): 320. doi: 10.1186/s12879-023-08289-7 . PMC 10173209 . PMID 37170214.
- 1 2 3 Khanlari M, Ramos JC, Sanchez SP, Cho-Vega JH, Amador A, Campuzano-Zuluaga G, Vega F, Chapman JR (July 2018). "Adult T-cell leukemia/lymphoma can be indistinguishable from other more common T-cell lymphomas. The University of Miami experience with a large cohort of cases". Modern Pathology. 31 (7): 1046–1063. doi:10.1038/s41379-018-0037-3. PMC 6931282 . PMID 29449683.
- ↑ Rezk SA, Zhao X, Weiss LM (June 2018). "Epstein–Barr virus–associated lymphoid proliferations, a 2018 update". Human Pathology. 79: 18–41. doi:10.1016/j.humpath.2018.05.020. PMID 29885408. S2CID 47010934.
- 1 2 Anagnostopoulos I, Hummel M, Tiemann M, Korbjuhn P, Parwaresch MR, Stein H (October 1994). "Frequent presence of latent Epstein-Barr virus infection in lymphoepithelioid cell lymphoma (Lennert's lymphoma)". Histopathology. 25 (4): 331–7. doi:10.1111/j.1365-2559.1994.tb01351.x. PMID 7835838.
- ↑ Yamashita Y, Nakamura S, Kagami Y, Hasegawa Y, Kojima H, Nagasawa T, Mori N (December 2000). "Lennert's lymphoma: a variant of cytotoxic T-cell lymphoma?". The American Journal of Surgical Pathology. 24 (12): 1627–33. doi:10.1097/00000478-200012000-00006. PMID 11117783.
- 1 2 3 Jeon YK (July 2016). "Clinicopathological analysis of a case series of peripheral T-cell lymphomas, not otherwise specified, of lymphoepithelioid variant (Lennert's lymphoma). A Central European single-center study-reply". Human Pathology. 53: 194–5. doi:10.1016/j.humpath.2015.12.034. PMID 27016487.
- 1 2 Cho U, Park G, Kim JA, Im S (February 2021). "Lymphoepithelioid variant of peripheral T cell lymphoma (Lennert lymphoma): Cytologic and histologic features". Diagnostic Cytopathology. 49 (2): 322–324. doi:10.1002/dc.24552. PMID 32745376.
- ↑ Stonesifer KJ, Benson NA, Ryden SE, Pawliger DF, Braylan RC (August 1986). "The malignant cells in a Lennert's lymphoma are T lymphocytes with a mature helper surface phenotype. A multiparameter flow cytometric analysis". Blood. 68 (2): 426–9. doi:10.1182/blood.V68.2.426.426. PMID 2942199.