
Fragile X syndrome (FXS) is a genetic disorder characterized by mild-to-moderate intellectual disability. The average IQ in males with FXS is under 55, while about two thirds of affected females are intellectually disabled. Physical features may include a long and narrow face, large ears, flexible fingers, and large testicles. About a third of those affected have features of autism such as problems with social interactions and delayed speech. Hyperactivity is common, and seizures occur in about 10%. Males are usually more affected than females.
Autism spectrum disorder (ASD) is a neurodevelopmental disorder that begins in early childhood, persists throughout adulthood, and affects two crucial areas of development: social communication and restricted, repetitive patterns of behavior. There are many conditions comorbid to autism spectrum disorder such as attention-deficit hyperactivity disorder and epilepsy.
Smith–Magenis syndrome (SMS), also known as 17p- syndrome, is a microdeletion syndrome characterized by an abnormality in the short (p) arm of chromosome 17. It has features including intellectual disability, facial abnormalities, difficulty sleeping, and numerous behavioral problems such as self-harm. Smith–Magenis syndrome affects an estimated between 1 in 15,000 to 1 in 25,000 individuals.
Neurodevelopmental disorders are a group of conditions that begin to emerge during childhood. According to the American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, (DSM-5) published in 2013, these conditions generally appear in early childhood, usually before children start school, and can persist into adulthood. The key characteristic of all these disorders is that they negatively impact a person's functioning in one or more domains of life depending on the disorder and deficits it has caused. All of these disorders and their levels of impairment exist on a spectrum, and affected individuals can experience varying degrees of symptoms and deficits, despite having the same diagnosis.

The heritability of autism is the proportion of differences in expression of autism that can be explained by genetic variation; if the heritability of a condition is high, then the condition is considered to be primarily genetic. Autism has a strong genetic basis. Although the genetics of autism are complex, autism spectrum disorder (ASD) is explained more by multigene effects than by rare mutations with large effects.

Medical genetics is the branch of medicine that involves the diagnosis and management of hereditary disorders. Medical genetics differs from human genetics in that human genetics is a field of scientific research that may or may not apply to medicine, while medical genetics refers to the application of genetics to medical care. For example, research on the causes and inheritance of genetic disorders would be considered within both human genetics and medical genetics, while the diagnosis, management, and counselling people with genetic disorders would be considered part of medical genetics.

XXYY syndrome is a sex chromosome anomaly in which males have 2 extra chromosomes, one X and one Y chromosome. Human cells usually contain two sex chromosomes, one from the mother and one from the father. Usually, females have two X chromosomes (XX) and males have one X and one Y chromosome (XY). The appearance of at least one Y chromosome with a properly functioning SRY gene makes a male. Therefore, humans with XXYY are genotypically male. Males with XXYY syndrome have 48 chromosomes instead of the typical 46. This is why XXYY syndrome is sometimes written as 48, XXYY syndrome or 48, XXYY. It affects an estimated one in every 18,000–40,000 male births.

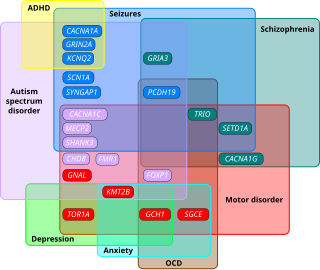
22q13 deletion syndrome, known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.
The Olduvai domain, known until 2018 as DUF1220 and the NBPF repeat, is a protein domain that shows a striking human lineage-specific (HLS) increase in copy number and appears to be involved in human brain evolution. The protein domain has also been linked to several neurogenetic disorders such as schizophrenia and increased severity of autism. In 2018, it was named by its discoverers after Olduvai Gorge in Tanzania, one of the most important archaeological sites for early humans, to reflect data indicating its role in human brain size and evolution.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.
Potocki–Lupski syndrome (PTLS), also known as dup(17)p11.2p11.2 syndrome, trisomy 17p11.2 or duplication 17p11.2 syndrome, is a contiguous gene syndrome involving the microduplication of band 11.2 on the short arm of human chromosome 17 (17p11.2). The duplication was first described as a case study in 1996. In 2000, the first study of the disease was released, and in 2007, enough patients had been gathered to complete a comprehensive study and give it a detailed clinical description. PTLS is named for two researchers involved in the latter phases, Drs. Lorraine Potocki and James R. Lupski of Baylor College of Medicine.

Pitt–Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, epilepsy, distinctive facial features, and possible intermittent hyperventilation followed by apnea. Pitt–Hopkins syndrome can be marked by intellectual disabilities as well as problems with socializing. It is part of the clinical spectrum of Rett-like syndromes.
1q21.1 deletion syndrome is a rare aberration of chromosome 1. A human cell has one pair of identical chromosomes on chromosome 1. With the 1q21.1 deletion syndrome, one chromosome of the pair is not complete, because a part of the sequence of the chromosome is missing. One chromosome has the normal length and the other is too short.

1q21.1 duplication syndrome or 1q21.1 (recurrent) microduplication is a rare aberration of chromosome 1.
Autism spectrum disorder (ASD) refers to a variety of conditions typically identified by challenges with social skills, communication, speech, and repetitive sensory-motor behaviors. The 11th International Classification of Diseases (ICD-11), released in January 2021, characterizes ASD by the associated deficits in the ability to initiate and sustain two-way social communication and restricted or repetitive behavior unusual for the individual's age or situation. Although linked with early childhood, the symptoms can appear later as well. Symptoms can be detected before the age of two and experienced practitioners can give a reliable diagnosis by that age. However, official diagnosis may not occur until much older, even well into adulthood. There is a large degree of variation in how much support a person with ASD needs in day-to-day life. This can be classified by a further diagnosis of ASD level 1, level 2, or level 3. Of these, ASD level 3 describes people requiring very substantial support and who experience more severe symptoms. ASD-related deficits in nonverbal and verbal social skills can result in impediments in personal, family, social, educational, and occupational situations. This disorder tends to have a strong correlation with genetics along with other factors. More research is identifying ways in which epigenetics is linked to autism. Epigenetics generally refers to the ways in which chromatin structure is altered to affect gene expression. Mechanisms such as cytosine regulation and post-translational modifications of histones. Of the 215 genes contributing, to some extent in ASD, 42 have been found to be involved in epigenetic modification of gene expression. Some examples of ASD signs are specific or repeated behaviors, enhanced sensitivity to materials, being upset by changes in routine, appearing to show reduced interest in others, avoiding eye contact and limitations in social situations, as well as verbal communication. When social interaction becomes more important, some whose condition might have been overlooked suffer social and other exclusion and are more likely to have coexisting mental and physical conditions. Long-term problems include difficulties in daily living such as managing schedules, hypersensitivities, initiating and sustaining relationships, and maintaining jobs.
Burnside–Butler syndrome is a name that has been applied to the effects of microdeletion of DNA sequences involving four neurodevelopmental genes. Varying developmental and psychiatric disorders have been attributed to the microdeletion; however, the great majority of people with the deletion do not have any clinical features associated with it. More studies are needed to delineate the range of clinical presentation.
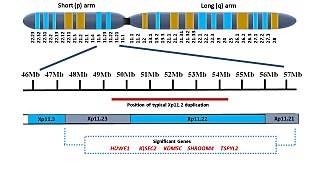
Xp11.2 duplication is a genomic variation marked by the duplication of an X chromosome region on the short arm p at position 11.2, defined by standard karyotyping (G-banding). This gene-rich, rearrangement prone region can be further divided into three loci - Xp11.21, Xp11.22 and Xp11.23. The duplication could involve any combination of these three loci. While the length of the duplication can vary from 0.5Mb to 55 Mb, most duplications measure about 4.5Mb and typically occur in the region of 11.22-11.23. Most affected females show preferential activation of the duplicated X chromosome. Features of affected individuals vary significantly, even among members of the same family. The Xp11.2 duplication can be 'silent' - presenting no obvious symptoms in carriers - which is known from the asymptomatic parents of affected children carrying the duplication. The common symptoms include intellectual disabilities, speech delay and learning difficulties, while in rare cases, children have seizures and a recognizable brain wave pattern when assessed by EEG (electroencephalography).

17q12 microdeletion syndrome, also known as 17q12 deletion syndrome, is a rare chromosomal anomaly caused by the deletion of a small amount of material from a region in the long arm of chromosome 17. It is typified by deletion of the HNF1B gene, resulting in kidney abnormalities and renal cysts and diabetes syndrome. It also has neurocognitive effects, and has been implicated as a genetic factor for autism and schizophrenia.

DiGeorge syndrome, also known as 22q11.2 deletion syndrome, is a syndrome caused by a microdeletion on the long arm of chromosome 22. While the symptoms can vary, they often include congenital heart problems, specific facial features, frequent infections, developmental disability, intellectual disability and cleft palate. Associated conditions include kidney problems, schizophrenia, hearing loss and autoimmune disorders such as rheumatoid arthritis or Graves' disease.
16p11.2 deletion syndrome is a rare genetic condition caused by microdeletion on the short arm of chromosome 16. Most affected individuals experience global developmental delay and intellectual disability, as well as childhood-onset obesity. 16p11.2 deletion is estimated to account for approximately 1% of autism spectrum disorder cases.
