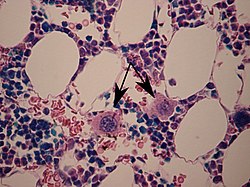
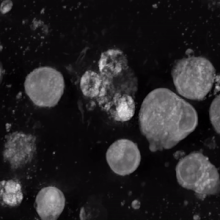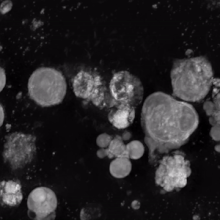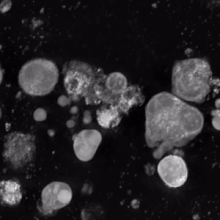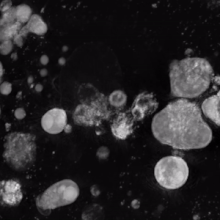
Haematopoiesis is the formation of blood cellular components. All cellular blood components are derived from haematopoietic stem cells. In a healthy adult human, roughly ten billion to a hundred billion new blood cells are produced per day, in order to maintain steady state levels in the peripheral circulation.

A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, and as a result, do not develop into healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include fatigue, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.

Fanconi anemia (FA) is a rare, AR, genetic disease resulting in impaired response to DNA damage in the FA/BRCA pathway. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), MDS, and liver tumors. 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity. 60% of FA is FANC-A, 16q24.3, which has later onset bone marrow failure.
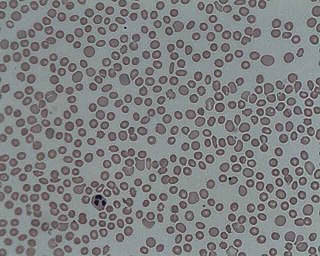
In hematology, thrombocytopenia is a condition characterized by abnormally low levels of platelets in the blood. Low levels of platelets in turn may lead to prolonged or excessive bleeding. It is the most common coagulation disorder among intensive care patients and is seen in a fifth of medical patients and a third of surgical patients.

Thrombopoietin (THPO) also known as megakaryocyte growth and development factor (MGDF) is a protein that in humans is encoded by the THPO gene.
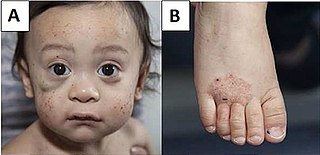
Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive disease characterized by eczema, thrombocytopenia, immune deficiency, and bloody diarrhea. It is also sometimes called the eczema-thrombocytopenia-immunodeficiency syndrome in keeping with Aldrich's original description in 1954. The WAS-related disorders of X-linked thrombocytopenia (XLT) and X-linked congenital neutropenia (XLN) may present with similar but less severe symptoms and are caused by mutations of the same gene.
Hematopoietic stem cells (HSCs) are the stem cells that give rise to other blood cells. This process is called haematopoiesis. In vertebrates, the very first definitive HSCs arise from the ventral endothelial wall of the embryonic aorta within the (midgestational) aorta-gonad-mesonephros region, through a process known as endothelial-to-hematopoietic transition. In adults, haematopoiesis occurs in the red bone marrow, in the core of most bones. The red bone marrow is derived from the layer of the embryo called the mesoderm.
Primary myelofibrosis (PMF) is a rare bone marrow blood cancer. It is classified by the World Health Organization (WHO) as a type of myeloproliferative neoplasm, a group of cancers in which there is activation and growth of mutated cells in the bone marrow. This is most often associated with a somatic mutation in the JAK2, CALR, or MPL genes. In PMF, the bony aspects of bone marrow are remodeled in a process called osteosclerosis; in addition, fibroblast secrete collagen and reticulin proteins that are collectively referred to as (fibrosis). These two pathological processes compromise the normal function of bone marrow resulting in decreased production of blood cells such as erythrocytes, granulocytes and megakaryocytes, the latter cells responsible for the production of platelets.

Myeloproliferative neoplasms (MPNs) are a group of rare blood cancers in which excess red blood cells, white blood cells or platelets are produced in the bone marrow. Myelo refers to the bone marrow, proliferative describes the rapid growth of blood cells and neoplasm describes that growth as abnormal and uncontrolled.
Myelokathexis is a congenital disorder of the white blood cells that causes severe, chronic leukopenia and neutropenia. The disorder is believed to be inherited in an autosomal dominant manner. Myelokathexis refers to retention (kathexis) of neutrophils in the bone marrow (myelo). The disorder shows prominent neutrophil morphologic abnormalities.
Mean platelet volume (MPV) is a machine-calculated measurement of the average size of platelets found in blood and is typically included in blood tests as part of the CBC. Since the average platelet size is larger when the body is producing increased numbers of platelets, the MPV test results can be used to make inferences about platelet production in bone marrow or platelet destruction problems.
Hemopoietic growth factors regulate the differentiation and proliferation of particular progenitor cells. Made available through recombinant DNA technology, they hold tremendous potential for medical uses when a person's natural ability to form blood cells is diminished or defective. Recombinant erythropoietin (EPO) is very effective in treating the diminished red blood cell production that accompanies end-stage kidney disease. Erythropoietin is a sialoglycoprotein hormone produced by peritubular cells of kidney.

GATA-binding factor 1 or GATA-1 is the founding member of the GATA family of transcription factors. This protein is widely expressed throughout vertebrate species. In humans and mice, it is encoded by the GATA1 and Gata1 genes, respectively. These genes are located on the X chromosome in both species.

Gray platelet syndrome (GPS), or platelet alpha-granule deficiency, is a rare congenital autosomal recessive bleeding disorder caused by a reduction or absence of alpha-granules in blood platelets, and the release of proteins normally contained in these granules into the marrow, causing myelofibrosis. The name derives from the initial observation of gray appearance of platelets with a paucity of granules on blood films from a patient with a lifelong bleeding disorder.
The Harrington–Hollingsworth experiment was an experiment that established the autoimmune nature of the blood disorder immune thrombocytopenic purpura. It was performed in 1950 by the academic staff of Barnes-Jewish Hospital in St. Louis, Missouri.

The thrombopoietin receptor also known as the myeloproliferative leukemia protein or CD110 is a protein that in humans is encoded by the MPL oncogene.

CFU-GEMM is a colony forming unit that generates myeloid cells. CFU-GEMM cells are the oligopotential progenitor cells for myeloid cells; they are thus also called common myeloid progenitor cells or myeloid stem cells. "GEMM" stands for granulocyte, erythrocyte, monocyte, megakaryocyte.
Congenital amegakaryocytic thrombocytopenia (CAMT) is a rare autosomal recessive bone marrow failure syndrome characterized by severe thrombocytopenia, which can progress to aplastic anemia and leukemia. CAMT usually manifests as thrombocytopenia in the initial month of life or in the fetal phase. Typically CAMPT presents with petechiae, cerebral bleeds, recurrent rectal bleeding, or pulmonary hemorrhage.

Thrombopoiesis is the formation of thrombocytes in the bone marrow. Thrombopoietin is the main regulator of thrombopoiesis. Thrombopoietin affects most aspects of the production of platelets. This includes self-renewal and expansion of hematopoietic stem cells, stimulating the increase of megakaryocyte progenitor cells, and supporting these cells so they mature to become platelet-producing cells. The process of Thrombopoiesis is caused by the breakdown of proplatelets. During the process almost all of the membranes, organelles, granules, and soluble macromolecules in the cytoplasm are being consumed. Apoptosis also plays a role in the final stages of thrombopoiesis by letting proplatelet processes to occur from the cytoskeleton of actin.

William Vainchenker, born on 16 December 1947, is a French medical doctor and researcher. He is considered a specialist in hematopoiesis.