In hematology, thrombocytopenia is a condition characterized by abnormally low levels of platelets (also known as thrombocytes) in the blood.[2] Low levels of platelets in turn may lead to prolonged or excessive bleeding. It is the most common coagulation disorder among intensive care patients and is seen in a fifth of medical patients and a third of surgical patients.[3]
A normal human platelet count ranges from 150,000 to 450,000 platelets/microliter (μL) of blood.[4] Values outside this range do not necessarily indicate disease. One common definition of thrombocytopenia requiring emergency treatment is a platelet count below 50,000/μL.[5] Thrombocytopenia can be contrasted with the conditions associated with an abnormally high level of platelets in the blood – thrombocythemia (when the cause is unknown), and thrombocytosis (when the cause is known).[6][7]
Signs and symptoms
Petechiae on the lower leg from thrombocytopeniaRight upper limb with purpura caused by thrombocytopenia in person with septic shock
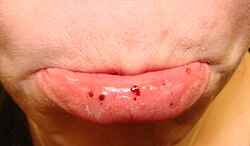
Thrombocytopenia usually has no symptoms and is picked up on a routine complete blood count. Some individuals with thrombocytopenia may experience external bleeding, such as nosebleeds or bleeding gums. Some women may have heavier or longer periods or breakthrough bleeding. Bruising, particularly purpura in the forearms and petechiae in the feet, legs, and mucous membranes, may be caused by spontaneous bleeding under the skin.[8][9]
Eliciting a full medical history is vital to ensure the low platelet count is not secondary to another disorder. Ensuring that the other blood cell types, such as red blood cells and white blood cells, are not also suppressed, is also important.[8] Painless, round, and pinpoint (1 to 3mm in diameter) petechiae usually appear and fade, and sometimes group to form ecchymoses. Larger than petechiae, ecchymoses are purple, blue, or yellow-green areas of skin that vary in size and shape. They can occur anywhere on the body.[8]
A person with this disease may also complain of malaise, fatigue, and general weakness (with or without accompanying blood loss). Acquired thrombocytopenia may be associated with the use of certain drugs. Inspection typically reveals evidence of bleeding (petechiae or ecchymoses), along with slow, continuous bleeding from any injuries or wounds. Adults may have large, blood-filled bullae in the mouth.[10] If the person's platelet count is between 30,000 and 50,000/μL, bruising with minor trauma may be expected; if it is between 15,000 and 30,000/μL, spontaneous bruising will be seen (mostly on the arms and legs).[11]
Causes
Thrombocytopenia can be inherited or acquired.[12] There are many causes.
Decreased production
Abnormally low platelet production may be caused by:[13]
Laboratory error, possibly due to the anticoagulant EDTA in CBC specimen tubes; a citrated platelet count is a useful follow-up study[17][additional citation(s) needed]
Cyclic thrombocytopenia, in which platelet counts vary in a cyclic pattern[22]
Diagnosis
Laboratory tests for thrombocytopenia might include full blood count, liver enzymes, kidney function, vitamin B12 levels, folic acid levels, erythrocyte sedimentation rate, and peripheral blood smear. If the cause for the low platelet count remains unclear, a bone marrow biopsy is usually recommended to differentiate cases of decreased platelet production from cases of peripheral platelet destruction.[23]
Thrombocytopenia in hospitalized alcoholics may be caused by spleen enlargement, folate deficiency, and most frequently, the direct toxic effect of alcohol on production, survival time, and function of platelets.[24] Platelet count begins to rise after 2 to 5 days' abstinence from alcohol. The condition is generally benign, and clinically significant hemorrhage is rare.[citation needed]
In severe thrombocytopenia, a bone marrow study can determine the number, size, and maturity of the megakaryocytes. This information may identify ineffective platelet production as the cause of thrombocytopenia and rule out a malignant disease process at the same time.[25]
Treatment
Treatment is guided by the severity and specific cause of the disease. Treatment focuses on eliminating the underlying problem, whether that means discontinuing drugs suspected to cause it or treating underlying sepsis. Diagnosis and treatment of serious thrombocytopenia is usually directed by a hematologist. Corticosteroids may be used to increase platelet production. Lithium carbonate or folate may also be used to stimulate platelet production in the bone marrow.[26]
Platelet transfusions
Platelet transfusions may be suggested for people who have a low platelet count due to thrombocytopenia.[27]
Thrombotic thrombocytopenic purpura
Treatment of thrombotic thrombocytopenic purpura (TTP) is a medical emergency, since the associated hemolytic anemia and platelet activation can lead to kidney failure and changes in the level of consciousness. Treatment of TTP was revolutionized in the 1980s with the application of plasmapheresis. According to the Furlan–Tsai hypothesis,[28] this treatment works by removing antibodies against the von Willebrand factor-cleaving proteaseADAMTS-13. The plasmapheresis procedure also adds active ADAMTS-13 protease proteins to the patient, restoring a normal level of von Willebrand factor multimers. Patients with persistent antibodies against ADAMTS-13 do not always manifest TTP, and these antibodies alone are not sufficient to explain how plasmapheresis treats TTP.[29]
Many cases of immune thrombocytopenic purpura (ITP), also known as idiopathic thrombocytopenic purpura, can be left untreated, and spontaneous remission (especially in children) is not uncommon. However, counts under 50,000/μL are usually monitored with regular blood tests, and those with counts under 10,000/μL are usually treated, as the risk of serious spontaneous bleeding is high with such low platelet counts. Any patient experiencing severe bleeding symptoms is also usually treated. The threshold for treating ITP has decreased since the 1990s; hematologists recognize that patients rarely spontaneously bleed with platelet counts greater than 10,000/μL, although exceptions to this observation have been documented.[30][31]
Thrombopoetin analogues have been tested extensively for the treatment of ITP. These agents had previously shown promise, but had been found to stimulate antibodies against endogenousthrombopoietin or lead to thrombosis. Romiplostim (trade name Nplate, formerly AMG 531) was found to be safe and effective for the treatment of ITP in refractory patients, especially those who relapsed following splenectomy.[32]
Discontinuation of heparin is critical in a case of heparin-induced thrombocytopenia (HIT). Beyond that, however, clinicians generally treat to avoid thrombosis.[33] Treatment may include a direct thrombin inhibitor, such as lepirudin or argatroban. Other "blood thinners" sometimes used in this setting include bivalirudin and fondaparinux. Platelet transfusions are not routinely used to treat HIT because thrombosis, not bleeding, is the primary problem.[34]Warfarin is not recommended until platelets have normalized.[34]
Congenital amegakaryocytic thrombocytopenia
Bone marrow/stem cell transplants are the only known cures for this genetic disease. Frequent platelet transfusions are required to keep the patient from bleeding to death before the transplant can be performed, although this is not always the case.[35]
Thrombocytopenia affects a few newborns, and its prevalence in neonatal intensive care units is high. Normally, it is mild and resolves without consequences. Most cases affect preterm birth infants and result from placental insufficiency and/or fetal hypoxia. Other causes, such as alloimmunity, genetics, autoimmunity, and infection, are less frequent.[37]
123"Thrombocytopenia". National Heart, Lung, and Blood Institute (NHLBI). U.S. Department of Health and Human Services. Archived from the original on 25 November 2020. Retrieved 4 January 2018.
↑Marini JJ, Dries DJ (2019). Critical care medicine: the essentials and more. Philadelphia: Wolters Kluwer. ISBN978-1-4963-0291-5. OCLC1060947164.
↑"Platelet count". MedlinePlus Medical Encyclopedia. U.S. National Library of Medicine. Archived from the original on 2015-04-05. Retrieved 2015-05-01.
↑"What Is Thrombocytopenia?". National Heart, Lung, and Blood Institute (NHLBI). U.S. Department of Health and Human Services. Archived from the original on 2015-05-18. Retrieved 2015-05-01.
↑"Thrombocythemia and Thrombocytosis". National Heart, Lung, and Blood Institute (NHLBI). U.S. Department of Health and Human Services. Archived from the original on 14 June 2019. Retrieved 5 August 2020.
↑"What Causes Thrombocytopenia?". National Heart, Lung, and Blood Institute (NHLBI). U.S. Department of Health and Human Services. Archived from the original on 2 December 2014. Retrieved 4 December 2014.
↑Almazni I, Stapley R, Morgan NV (2019) Inherited Thrombocytopenia: Update on genes and genetic variants which may be associated With bleeding. Front Cardiovasc Med
↑"How Is Thrombocytopenia Diagnosed?". National Heart, Lung, and Blood Institute (NHLBI). U.S. Department of Health and Human Services. Archived from the original on 2015-05-25. Retrieved 2015-05-19.
1234567Guida JD, Kunig AM, Leef KH, McKenzie SE, Paul DA (June 2003). "Platelet count and sepsis in very low birth weight neonates: is there an organism-specific response?". Pediatrics. 111 (6 Pt 1): 1411–1415. doi:10.1542/peds.111.6.1411. PMID12777561.
12Pacifico L, Chiesa C, Mirabella S, Panero A, Midulla M (March 1987). "Early-onset Pseudomonas aeruginosa sepsis and Yersinia enterocolitica neonatal infection: a unique combination in a preterm infant". European Journal of Pediatrics. 146 (2): 192–193. doi:10.1007/BF02343233. PMID3569360. S2CID20198866.
↑Rempen A, Martius J, Hartmann AA, Wecker I (1987). "Transmission rate of Ureaplasma urealyticum, Mycoplasma spp., Gardnerella vaginalis, B-streptococci, Candida spp. and Chlamydia trachomatis from the mother to the newborn". Archives of Gynecology and Obstetrics. 241 (3): 165–170. doi:10.1007/BF00931313. PMID3324978. S2CID11251976.
↑Kotiw M, Zhang GW, Daggard G, Reiss-Levy E, Tapsall JW, Numa A (2003). "Late-onset and recurrent neonatal Group B streptococcal disease associated with breast-milk transmission". Pediatric and Developmental Pathology. 6 (3): 251–256. doi:10.1007/s10024-001-0276-y. PMID12687430. S2CID20696142.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.