Thrombophilia (sometimes called hypercoagulability or a prothrombotic state) is an abnormality of blood coagulation that increases the risk of thrombosis (blood clots in blood vessels).[1][2] Such abnormalities can be identified in 50% of people who have an episode of thrombosis (such as deep vein thrombosis in the leg) that was not provoked by other causes.[3] A significant proportion of the population has a detectable thrombophilic abnormality, but most of these develop thrombosis only in the presence of an additional risk factor.[2]
There is no specific treatment for most thrombophilias, but recurrent episodes of thrombosis may be an indication for long-term preventive anticoagulation.[2] The first major form of thrombophilia to be identified by medical science, antithrombin deficiency, was identified in 1965, while the most common abnormalities (including factor V Leiden) were described in the 1990s.[4][5]
Signs and symptoms
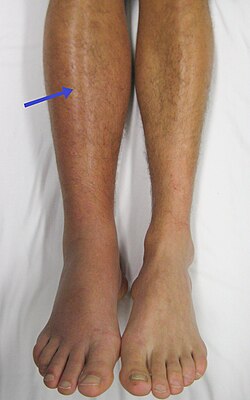
A right-sided acute deep vein thrombosis (to the left in the image). The leg is swollen and red due to venous outflow obstruction.
Protein C deficiency may cause purpura fulminans, a severe clotting disorder in the newborn that leads to both tissue death and bleeding into the skin and other organs. The condition has also been described in adults. Protein C and protein S deficiency have also been associated with an increased risk of skin necrosis on commencing anticoagulant treatment with warfarin or related drugs.[2][12]
Causes
Thrombophilia can be congenital or acquired. Congenital thrombophilia refers to inborn conditions (and usually hereditary, in which case "hereditary thrombophilia" may be used) that increase the tendency to develop thrombosis, while, on the other hand, acquired thrombophilia refers to conditions that arise later in life.
Congenital
The most common types of congenital thrombophilia are those that arise as a result of overactivity of coagulation factors; hence they are considered "gain-of-function" alterations.[13] They are relatively mild in the usual heterozygous state, and are therefore classified as "type II" defects.[14][15] The most common ones are factor V Leiden (a mutation in the F5 gene at position 1691) and prothrombin G20210A, a mutation in prothrombin (at position 20210 in the 3' untranslated region of the gene).[1][16] Compound heterozygotes and homozygotes, while rare, are at significant risk of thrombosis.[13]
The rare forms of congenital thrombophilia are typically caused by a deficiency of natural anticoagulants. They are classified as "type I" and are more severe in their propensity to cause thrombosis.[14] The main ones are antithrombin III deficiency, protein C deficiency and protein S deficiency.[1][16] Milder rare congenital thrombophilias are factor XIII mutation[16] and familial dysfibrinogenemia (an abnormal fibrinogen).[16] It is unclear whether congenital disorders of fibrinolysis (the system that destroys clots) are major contributors to thrombosis risk.[14] Congenital deficiency of plasminogen, for instance, mainly causes eye symptoms and sometimes problems in other organs, but the link with thrombosis has been more uncertain.[17]
Blood group determines thrombosis risk to a significant extent. Those with blood groups other than typeO are at a 2- to 4-fold relative risk. O blood group is associated with reduced levels of von Willebrand factor — because of increased clearance — and factor VIII, which is related to thrombotic risk .[5]
Heparin-induced thrombocytopenia (HIT) is due to an immune system reaction against the anticoagulant drug heparin (or its derivatives).[1] Though it is named for associated low platelet counts, HIT is strongly associated with risk of venous and arterial thrombosis.[19]Paroxysmal nocturnal hemoglobinuria (PNH) is a rare condition resulting from acquired alterations in the PIGA gene, which plays a role in the protection of blood cells from the complement system. PNH increases the risk of venous thrombosis but is also associated with hemolytic anemia (anemia resulting from destruction of red blood cells).[20] Both HIT and PNH require particular treatment.[19][20]
Hematologic conditions associated with sluggish blood flow can increase risk for thrombosis. For example, sickle-cell disease (caused by mutations of hemoglobin) is regarded as a mild prothrombotic state induced by impaired flow.[1] Similarly, myeloproliferative disorders, in which the bone marrow produces too many blood cells, predispose to thrombosis, particularly in polycythemia vera (excess red blood cells) and essential thrombocytosis (excess platelets). Again, these conditions usually warrant specific treatment when identified.[21]
Cancer, particularly when metastatic (spread to other places in the body), is a recognised risk factor for thrombosis.[2][16] A number of mechanisms have been proposed, such as activation of the coagulation system by cancer cells or secretion of procoagulant substances. Furthermore, particular cancer treatments (such as the use of central venous catheters for chemotherapy) may increase the risk of thrombosis further.[22]
Nephrotic syndrome, in which protein from the bloodstream is released into the urine due to kidney diseases, can predispose to thrombosis;[1] this is particularly the case in more severe cases (as indicated by blood levels of albumin below 25g/L) and if the syndrome is caused by the condition membranous nephropathy.[23]Inflammatory bowel disease (ulcerative colitis and Crohn's disease) predispose to thrombosis, particularly when the disease is active. Various mechanisms have been proposed.[2][24]
Obesity has long been regarded as a risk factor for venous thrombosis. It more than doubles the risk in numerous studies, particularly in combination with the use of oral contraceptives or in the period after surgery. Various coagulation abnormalities have been described in the obese. Plasminogen activator inhibitor-1, an inhibitor of fibrinolysis, is present in higher levels in people with obesity. Obese people also have larger numbers of circulating microvesicles (fragments of damaged cells) that bear tissue factor. Platelet aggregation may be increased, and there are higher levels of coagulation proteins such as von Willebrand factor, fibrinogen, factor VII and factor VIII. Obesity also increases the risk of recurrence after an initial episode of thrombosis.[30]
There is an association between the blood levels of homocysteine and thrombosis,[16] although this has not been reported consistently in all studies.[5] Homocysteine levels are determined by mutations in the MTHFR and CBS genes, but also by levels of folic acid, vitamin B6 and vitamin B12, which depend on diet.[14]
The coagulation system, often described as a "cascade", consists of a group of proteins that interact in the formation of a fibrin-rich clot.
Thrombosis is a multifactorial problem because there are often multiple reasons why a person might develop thrombosis. These risk factors may include any combination of abnormalities in the blood vessel wall, abnormalities in the blood flow (as in immobilization), and abnormalities in the consistency of the blood. Thrombophilia is caused by abnormalities in blood consistency, which is determined by the levels of coagulation factors and other circulating blood proteins that participate in the "coagulation cascade".[16]
Normal coagulation is initiated by the release of tissue factor from damaged tissue. Tissue factor binds to circulating factor VIIa. The combination activates factor X to factor Xa and factor IX to factor IXa. Factor Xa (in the presence of factor V) activates prothrombin into thrombin. Thrombin is a central enzyme in the coagulation process: it generates fibrin from fibrinogen, and activates a number of other enzymes and cofactors (factor XIII, factor XI, factor V and factor VIII, TAFI) that enhance the fibrin clot.[14] The process is inhibited by TFPI (which inactivates the first step catalyzed by factor VIIa/tissue factor), antithrombin (which inactivates thrombin, factor IXa, Xa and XIa), protein C (which inhibits factors Va and VIIIa in the presence of protein S), and protein Z (which inhibits factor Xa).[14]
In thrombophilia, the balance between "procoagulant" and "anticoagulant" activity is disturbed. The severity of the imbalance determines the likelihood that someone develops thrombosis. Even small perturbances of proteins, such as the reduction of antithrombin to only 70–80% of the normal level, can increase the thrombosis risk; this is in contrast with hemophilia, which only arises if levels of coagulation factors are markedly decreased.[14]
In addition to its effects on thrombosis, hypercoagulable states may accelerate the development of atherosclerosis, the arterial disease that underlies myocardial infarction and other forms of cardiovascular disease.[31][32]
Diagnosis
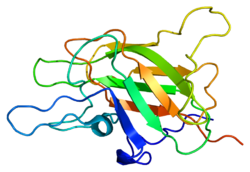
A mutation of coagulation factor V (schematic representation drawn here) is much more common in people with thrombosis than in those without, but is only regarded as a weak risk factor.
For hereditary cases, the patient must have at least two abnormal tests plus family history.
Screening
There are divergent views as to whether everyone with an unprovoked episode of thrombosis should be investigated for thrombophilia. Even those with a form of thrombophilia may not necessarily be at risk of further thrombosis, while recurrent thrombosis is more likely in those who have had previous thrombosis even in those who have no detectable thrombophilic abnormalities.[8][12][33] Recurrent thromboembolism, or thrombosis in unusual sites (e.g. the hepatic vein in Budd-Chiari syndrome), is a generally accepted indication for screening. It is more likely to be cost-effective in people with a strong personal or family history of thrombosis.[34] In contrast, the combination of thrombophilia with other risk factors may provide an indication for preventive treatment, which is why thrombophilia testing may be performed even in those who would not meet the strict criteria for these tests.[33] Searching for a coagulation abnormality is not normally undertaken in patients in whom thrombosis has an obvious trigger. For example, if the thrombosis is due to immobilization after recent orthopedic surgery, it is regarded as "provoked" by the immobilization and the surgery and it is less likely that investigations will yield clinically important results.[12][33]
When venous thromboembolism occurs when a patient is experiencing transient major risk factors such as prolonged immobility, surgery, or trauma, testing for thrombophilia is not appropriate because the outcome of the test would not change a patient's indicated treatment.[35][36] In 2013, the American Society of Hematology, as part of recommendations in the Choosing Wisely campaign, cautioned against overuse of thrombophilia screening; false positive results of testing would lead to people inappropriately being labeled as having thrombophilia, and being treated with anticoagulants without clinical need.[35] A 2016 study estimated that more than $1 million was wasted on inappropriate thrombophilia testing in a year at one academic medical center.[37]
In the United Kingdom, professional guidelines give specific indications for thrombophilia testing. It is recommended that testing be done only after appropriate counseling, and hence the investigations are usually not performed at the time when thrombosis is diagnosed but at a later time.[12] In particular situations, such as retinal vein thrombosis, testing is discouraged altogether because thrombophilia is not regarded as a major risk factor. In other rare conditions generally linked with hypercoagulability, such as cerebral venous thrombosis and portal vein thrombosis, there is insufficient data to state for certain whether thrombophilia screening is helpful, and decisions on thrombophilia screening in these conditions are therefore not regarded as evidence-based.[12] If cost-effectiveness (quality-adjusted life years in return for expenditure) is taken as a guide, it is generally unclear whether thrombophilia investigations justify the often high cost,[38] unless the testing is restricted to selected situations.[39]
In 2021, the American College of Chest Physicians offered one testing-related recommendation in its venous thromboembolism guidelines.[40] They recommended to consider positive D-dimer in the decision to continue or discontinue anticoagulation. Positive D-dimer may suggest that the ongoing thrombotic tendency has not fully resolved.
In 2023, the American Society of Hematology issued new guidelines for thrombophilia testing.[41] One departure from their previous guidelines relates to patients with nonsurgical major transient risk factors; testing may be appropriate. Thrombophilia testing after venous thromboembolism(VTE) provoked by surgery, on the other hand, is not recommended, because the risk of recurrence is low. Some experts argue that unprovoked VTE requires indefinite (lifelong) anticoagulation and therefore performing thrombophilia testing will not affect management. Nearly all recommendations in the guidelines were based on "very low certainty" evidence.[41]
Recurrent miscarriage is an indication for thrombophilia screening, particularly antiphospholipid antibodies (anti-cardiolipin IgG and IgM, as well as lupus anticoagulant), factor V Leiden and prothrombin mutation, activated protein C resistance and a general assessment of coagulation through an investigation known as thromboelastography.[11]
Women who are planning to use oral contraceptives do not benefit from routine screening for thrombophilias, as the absolute risk of thrombotic events is low.[41] If either the woman or a first-degree relative has had thrombosis, the risk of developing thrombosis is increased. Screening this selected group may be beneficial,[29] but even when negative may still indicate residual risk.[12] Professional guidelines therefore suggest that alternative forms of contraception be used rather than relying on screening.[12]
Thrombophilia screening in people with arterial thrombosis is generally regarded as unrewarding and is generally discouraged,[12] except possibly for unusually young patients (especially when precipitated by smoking or use of estrogen-containing hormonal contraceptives) and those in whom revascularization, such as coronary arterial bypass, fails because of rapid occlusion of the graft.[9]
Timing of Testing
Several thrombophilia assays can be impacted by the presence of anticoagulants. Therefore, most thrombophilia testing should be done after the patient has completed the initial treatment course of anticoagulation.[41] Efforts to remove direct oral anticoagulants using activated carbon[42] may prove helpful in this regard.
People considered to be at a high risk of repeated thrombosis due to thrombophilia are often advised to take warfarin for prolonged periods of time or even indefinitely.
There is no specific treatment for thrombophilia, unless it is caused by an underlying medical illness (such as nephrotic syndrome), where the treatment of the underlying disease is needed. In those with unprovoked and/or recurrent thrombosis, or those with a high-risk form of thrombophilia, the most important decision is whether to use anticoagulation medications, such as warfarin, on a long-term basis to reduce the risk of further episodes.[3] This risk needs to weighed against the risk that the treatment will cause significant bleeding, as the reported risk of major bleeding is over 3% per year, and 11% of those with major bleeding may die as a result.[3]
Apart from the abovementioned forms of thrombophilia, the risk of recurrence after an episode of thrombosis is determined by factors such as the extent and severity of the original thrombosis, whether it was provoked (such as by immobilization or pregnancy), the number of previous thrombotic events, male sex, the presence of an inferior vena cava filter, the presence of cancer, symptoms of post-thrombotic syndrome, and obesity.[3] These factors tend to be more important in the decision than the presence or absence of a detectable thrombophilia.[12][43]
Those with antiphospholipid syndrome may be offered long-term anticoagulation after a first unprovoked episode of thrombosis. The risk is determined by the subtype of antibody detected, by the antibody titer (amount of antibodies), whether multiple antibodies are detected, and whether it is detected repeatedly or only on a single occasion.[18]
Women with a thrombophilia who are contemplating pregnancy or are pregnant usually require alternatives to warfarin during pregnancy, especially in the first 13 weeks, when it may produce abnormalities in the unborn child. Low molecular weight heparin (LMWH, such as enoxaparin) is generally used as an alternative.[44] Warfarin and LMWH may safely be used in breastfeeding.[44]
When women experience recurrent pregnancy loss secondary to thrombophilia, some studies have suggested that low molecular weight heparin reduces the risk of miscarriage. When the results of all studies are analysed together, no statistically significant benefit could be demonstrated.[45]
Prognosis
In people without a detectable thrombophilia, the cumulative risk of developing thrombosis by the age of 60 is about 12%. About 60% of people who are deficient in antithrombin will have experienced thrombosis at least once by age 60, as will about 50% of people with protein C deficiency and about a third of those with protein S deficiency. People with activated protein C resistance (usually resulting from factor V Leiden), in contrast, have a slightly raised absolute risk of thrombosis, with 15% having had at least one thrombotic event by the age of sixty.[14] In general, men are more likely than women to experience repeated episodes of venous thrombosis.[5]
People with factor V Leiden are at a relatively low risk of thrombosis, but may develop thrombosis in the presence of an additional risk factor, such as immobilization. Most people with the prothrombin mutation (G20210A) never develop thrombosis.[14]
Epidemiology
The major ("type 1") thrombophilias are rare. Antithrombin deficiency is present in 0.2% of the general population and 0.5–7.5% of people with venous thrombosis. Protein C deficiency, too, is present in 0.2% of the population, and can be found in 2.5–6% of people with thrombosis. The exact prevalence of protein S deficiency in the population is unknown; it is found 1.3–5% of people with thrombosis.[14]
The minor ("type 2") thrombophilias are much more common. Factor V Leiden is present in 5% of the population of Northern European descent, but much rarer in those of Asian or African extraction. In people with thrombosis, 10% have factor V Leiden. In those who are referred for thrombophilia testing, 30–50% have the defect. The prothrombin mutation occurs at rates of 1–4% in the general population, 5–10% of people with thrombosis, and 15% of people referred for thrombophilia testing. Like factor V Leiden, this abnormality is uncommon in Africans and Asians.[14]
The exact prevalence of antiphospholipid syndrome is not well known, as different studies employ different definitions of the condition. Antiphospholipid antibodies are detected in 24% of those referred to thrombophilia testing.[18]
History
Rudolf Virchow, the German pathologist who distinguished the various causes of thrombosis, and whose work led to the development of thrombophilia as a concept
Antiphospholipid syndrome was described in full in the 1980s, after various previous reports of specific antibodies in people with systemic lupus erythematosus and thrombosis.[18][49] The syndrome is often attributed to the British rheumatologist Graham R.V. Hughes, and is often referred to as Hughes syndrome for that reason.[50]
The more common genetic thrombophilias were described in the 1990s. Many studies had previously indicated that many people with thrombosis showed resistance activated protein C. In 1994 a group in Leiden, The Netherlands, identified the most common underlying defect—a mutation in factor V that made it resistant to the action of activated protein C. The defect was called factor V Leiden, as genetic abnormalities are typically named after the place where they are discovered.[51] Two years later, the same group described a common mutation in the prothrombin gene that caused elevation of prothrombin levels and a mild increase in thrombosis risk.[4][5][52]
↑ Borissoff JI, Spronk HM, ten Cate H (2011). "The hemostatic system as a modulator of atherosclerosis". N. Engl. J. Med. 364 (18): 1746–60. doi:10.1056/NEJMra1011670. PMID21542745.
Chong LY, Fenu E, Stansby G, Hodgkinson S, Guideline Development G (2012). "Management of venous thromboembolic diseases and the role of thrombophilia testing: Summary of NICE guidance". BMJ. 344: e3979. doi:10.1136/bmj.e3979. PMID22740565. S2CID42342532.
↑ Exner T, Rigano J, Favaloro EJ (June 2020). "The effect of DOACs on laboratory tests and their removal by activated carbon to limit interference in functional assays". Int J Lab Hematol. 42 (Suppl 1): 41–48. doi:10.1111/ijlh.13196. PMID32543072.
↑ Comp PC, Esmon CT (December 1984). "Recurrent venous thromboembolism in patients with a partial deficiency of protein S". N. Engl. J. Med. 311 (24): 1525–8. doi:10.1056/NEJM198412133112401. PMID6239102.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.