The medical management of individuals with hemophilia A frequently entails the administration of factor VIII medication through slow intravenous injection. This intervention aims to address and preempt additional bleeding episodes in affected individuals.
Signs and symptoms
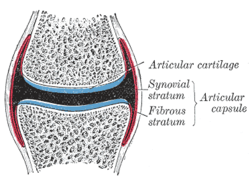
Joint capsule
Haemophilia A's phenotype has quite a wide range of symptoms encompassing both internal and external bleeding episodes. Individuals with more severe haemophilia tend to experience more intense and frequent bleeding, whereas those with mild haemophilia typically exhibit milder symptoms unless subjected to surgical procedures or significant trauma. Those with moderate haemophilia may display variable symptoms, falling within the spectrum between severe and mild forms.
One common early indicator of haemophilia is prolonged bleeding from venepuncture or heelpricks. These signs often prompt blood tests that confirm the presence of haemophilia.[5] In individuals, especially those with moderate or mild haemophilia, any form of trauma can trigger the first significant bleed. Haemophilia substantially elevates the risk of protracted bleeding from ordinary injuries, and in severe cases, bleeding can occur spontaneously without an apparent cause. Bleeding episodes can manifest anywhere in the body. Superficial bleeding resulting from abrasions or shallow lacerations may persist, with scabs easily breaking due to the deficiency of fibrin, potentially leading to re-bleeding.[1] While superficial bleeding poses challenges, more critical sites of bleeding include:[6]
One therapeutic conundrum is the development of inhibitor antibodies against factor VIII due to frequent infusions. These develop as the body recognises the infused factor VIII as foreign, as the body does not produce its own copy. In these individuals, activated factor VII, a protein in the extrinsic pathway of the coagulation cascade, can be infused as a treatment for haemorrhage in individuals with haemophilia and antibodies against replacement factor VIII.[1][7]
Oral Manifestations
The oral manifestations are characterized by frequent bleeding of multiple sites, frequently seen as gingival and postextraction haemorrhages. The symptoms depend on the severity of haemophilia. In the case of severe haemophilia, patients may complain of multiple oral bleeding episodes throughout their life. Haemophilia patients are considered to be a special group of patients as routinely done procedures may be fatal in them. It was seen that almost 14% of all haemophilia patients and 30% of cases with a mild type of haemophilia have been diagnosed early following an episode of severe oral bleeding, of which the most common sites were the labial frenum and the tongue.[8]
Genetics
X linked recessive inheritance
Haemophilia A is inherited as an X-linkedrecessive trait. It occurs in males and in homozygous females (which is only possible in the daughters of a haemophilic male and a carrier or haemophiliac female[9]). However, mild haemophilia A is known to occur in heterozygous females due to X-inactivation, so it is recommended that levels of factor VIII and IX be measured in all known or potential carriers prior to surgery and in the event of clinically significant bleeding.[1][10]
About 5-10% of people with haemophilia A are affected because they make a dysfunctional version of the factor VIII protein, while the remainder are affected because they produce factor VIII in insufficient amounts (quantitative deficiency).[10] Of those who have severe deficiency (defined as <1% activity of factor VIII), 45-50% have the same mutation, an inversion within the factor VIII gene that results in total elimination of protein production.[10]
Since both forms of haemophilia can be caused by a variety of different mutations, initial diagnosis and classification is done by measurement of protein activity rather than by genetic tests, though genetic tests are recommended for testing of family members once a known case of haemophilia is identified.[1][10] Approximately 30% of patients have no family history; their disease is presumably caused by new mutations.[11]
Causes
Vaccine
In 2022, it was reported that a man in his 80s developed Acquired Haemophilia A (AHA) two weeks after he got an injection of COVID-19 mRNA vaccine. Several tests demonstrated an isolated prolonged activated partial thromboplastin time (aPTT) not corrected immediately and low clotting factor VIII (FVIII) activity (6.7percent). Researchers hypothesized that the mRNA vaccination induced an autoimmune response, considering vaccines can activate nonspecific antibodies and stimulate autoantibody production.[12]
AHA patients are mostly aged over 65, but it was reported that a 45-year-old woman who had no medical condition suffered from AHA two weeks after receiving COVID-19 mRNA booster injection. Researchers observed a drop in FVIII activity (0.9 percent), prolonged aPPT (75.1 seconds), and confirmed FVIII inhibitor (12.8 BU/mL), and could find no indication that genetic factors were responsible for. Their study suggested that young adults could suffer from AHA after receiving COVID-19 mRNA shots. [13]
In 2024, Korean researchers reported that a 56-year-old man who had medical conditions such as diabetes and dyslipidemia was diagnosed with AHA 15 days after receiving his second dose of COVID-19 mRNA vaccine. It was Korea's first case of post-vaccination AHA concerning COVID-19 vaccine. They argued that monitoring individuals after COVID-19 vaccination was essential for detecting rare adverse effects. [14]
Diagnosis
X-Ray- haemoarthritisHaemarthrosis on lateral view
The diagnosis of haemophilia A may be suspected as coagulation testing reveals an increased partial thromboplastin time (PTT) in the context of a normal prothrombin time (PT) and bleeding time. PTT tests are the first blood test done when haemophilia is indicated.[15] However, the diagnosis is made in the presence of very low levels of factor VIII. A family history is frequently present, although not essential. Recently, genetic testing has been made available to determine an individual's risk of attaining or passing on haemophilia. Diagnosis of haemophilia A also includes a severity level, which can range from mild to severe based on the amount of active and functioning factor VIII detected in the blood. Factor VIII levels do not typically change throughout an individual's lifetime. Severe haemophilia A is the most common severity, occurring in the majority of affected people. Individuals with mild haemophilia often experience few or no bleeding episodes except in the case of serious trauma (i.e. tooth extraction, or surgery).[1]
Severity
There are numerous different mutations which can cause haemophilia A, due to differences in changes to the factor VIII gene (and the resulting protein). Individuals with haemophilia often have some level of active clotting factor. Individuals with less than 1% active factor are classified as having severe haemophilia, those with 1–5% active factor have moderate haemophilia, and those with mild haemophilia have between 5–40% of normal levels of active clotting factor.[16]
Most individuals with severe haemophilia require regular supplementation with intravenousplasma concentrate factor VIII or efanesoctocog alfa, a von Willebrand factor (VWF) independent, recombinant DNA-derived Factor VIII (FVIII) concentrate shown to prevent bleeding in children and adults.[18][19] Treatment dosing and frequency of plasma concentrate Factor VIII may be variable and individually determined;[6] dosing of efanesoctocog alfa shown effective is an IV injection of once-weekly 50 IU per kilogram of body weight.
In children, an easily accessible intravenous port[20] may have to be inserted to minimise frequent traumatic intravenous cannulation. These devices have made prophylaxis in haemophilia much easier for families because the problems of finding a vein for infusion several times a week are eliminated. However, there are risks involved with their use, the most worrisome being that of infection, studies differ but some show an infection rate that is high.[21] These infections can usually be treated with intravenous antibiotics but sometimes the device must be removed,[22] also, there are other studies that show a risk of clots forming at the tip of the catheter, rendering it useless. Some individuals with severe haemophilia, and most with moderate and mild haemophilia, treat only as needed without a regular prophylactic schedule.[23] Mild haemophiliacs often manage their condition with desmopressin, a drug which releases stored factor VIII from blood vessel walls.[24]
Fitusiran (Qfitlia) was approved for medical use in the United States in March 2025.[25][26]
Dental considerations
If numbing is necessary for dental procedures, the nerve block (typically to the inferior alveolar nerve) should only be given after raising clotting factor levels by appropriate replacement therapy, as there is a risk of bleeding into the muscles along with potential airway compromise due to a haematoma in the retromolar or pterygoid space. The intraligamental technique or interosseous technique should be considered instead of the mandibular block. Articaine has been used as a buccal infiltration to anaesthetize the lower molar teeth. A lingual infiltration also requires appropriate factor replacement since the injection is into an area with a rich plexus of blood vessels and the needle is not adjacent to bone.[27]
Gene therapy
In December 2017, it was reported that doctors had used a new form of gene therapy to treat haemophilia A.[28][29][30] Current treatment efforts utilize adeno-associated virus (AAV) vectors, however recent studies have found lentiviral vectors (LV) as being a more effective alternative.[31][32]
A study published in July 2024, demonstrated that efanesoctocog alfa, a bioengineered human factor VIII recombinant protein, prophylaxis for children with severe hemophilia A could have therapeutic benefit leading to effective bleeding prevention.[34][35]
Prognosis
Two Dutch studies have followed haemophilia patients for a number of years.[36][37] Both studies found that viral infections were common in haemophiliacs due to the frequent blood transfusions which put them at risk of acquiring blood borne infections, such as HIV, hepatitis B and hepatitis C. In the latest study which followed patients from 1992 to 2001, the male life expectancy was 59 years. If cases with known viral infections were excluded, the life expectancy was 72, close to that of the general population. 26% of the cases died from AIDS and 22% from hepatitis C.[37] However, these statistics for prognosis are unreliable as there has been marked improvement of infection control and efficacy of anti-retroviral drugs since these studies were done.[citation needed]
Epidemiology
Haemophilia A occurs in approximately 1 in 5,000 males,[10] while the incidence of haemophilia B is 1 in 30,000 in the male population,[10] of these, 85% have haemophilia A and 15% have haemophilia B.[10]
↑Andrew Brewer, Maria Elvira Correa (May 2006). "Guildelines for Dental Treatment of Patients with Inherited Bleeding Disorders" (PDF). Treatment of Hemophilia. 40: 9 – via World Federation of Hemophilia (WFH).
12Plug, I.; Van Der Bom, J. G.; Peters, M.; Mauser-Bunschoten, E. P.; De Goede-Bolder, A.; Heijnen, L.; Smit, C.; Willemse, J.; Rosendaal, F. R. (2006-03-01). "Mortality and causes of death in patients with hemophilia, 1992–2001: a prospective cohort study1". Journal of Thrombosis and Haemostasis. 4 (3): 510–516. doi:10.1111/j.1538-7836.2006.01808.x. hdl:1887/5021. ISSN1538-7836. PMID16460432. S2CID13651790.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.