Kocher–Debré–Semelaigne syndrome (KDSS) is hypothyroidism in infancy or childhood characterised by lower extremity or generalized muscular hypertrophy, myxoedema, short stature, and cognitive impairment.

Glycogen storage disease type V, also known as McArdle's disease, is a metabolic disorder, one of the metabolic myopathies, more specifically a muscle glycogen storage disease, caused by a deficiency of myophosphorylase. Its incidence is reported as one in 100,000, roughly the same as glycogen storage disease type I.

A glycogen storage disease is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.

Fabry disease, also known as Anderson–Fabry disease, is a rare genetic disease that can affect many parts of the body, including the kidneys, heart, brain, and skin. Fabry disease is one of a group of conditions known as lysosomal storage diseases. The genetic mutation that causes Fabry disease interferes with the function of an enzyme that processes biomolecules known as sphingolipids, leading to these substances building up in the walls of blood vessels and other organs. It is inherited in an X-linked manner.

Alpha-mannosidosis is a lysosomal storage disorder, first described by Swedish physician Okerman in 1967. In humans it is known to be caused by an autosomal recessive genetic mutation in the gene MAN2B1, located on chromosome 19, affecting the production of the enzyme alpha-D-mannosidase, resulting in its deficiency. Consequently, if both parents are carriers, there will be a 25% chance with each pregnancy that the defective gene from both parents will be inherited, and the child will develop the disease. There is a two in three chance that unaffected siblings will be carriers. In livestock alpha-mannosidosis is caused by chronic poisoning with swainsonine from locoweed.

Glycogen storage disease type II(GSD-II), also called Pompe disease, and formerly known as GSD-IIa or Limb–girdle muscular dystrophy2V, is an autosomal recessive metabolic disorder which damages muscle and nerve cells throughout the body. It is caused by an accumulation of glycogen in the lysosome due to deficiency of the lysosomal acid alpha-glucosidase enzyme (GAA). The inability to breakdown glycogen within the lysosomes of cells leads to progressive muscle weakness throughout the body and affects various body tissues, particularly in the heart, skeletal muscles, liver and the nervous system.

Phosphofructokinase deficiency is a rare muscular metabolic disorder, with an autosomal recessive inheritance pattern. It is characterized as a deficiency in the Phosphofructokinase (PFK) enzyme throughout the body, including the skeletal muscles and red blood cells. Phosphofrucotkinase is an enzyme involved in the glycolytic process. The lack of PFK blocks the completion of the glycolytic pathway. Therefore, all products past the block would be deficient, including Adenosine triphosphate (ATP).
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are often referred to as congenital metabolic diseases or inherited metabolic disorders. Another term used to describe these disorders is "enzymopathies". This term was created following the study of biodynamic enzymology, a science based on the study of the enzymes and their products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene–one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.
In medicine, myopathy is a disease of the muscle in which the muscle fibers do not function properly. Myopathy means muscle disease. This meaning implies that the primary defect is within the muscle, as opposed to the nerves or elsewhere.
Hereditary inclusion body myopathies (HIBM) are a group of rare genetic disorders which have different symptoms. Generally, they are neuromuscular disorders characterized by muscle weakness developing in young adults. Hereditary inclusion body myopathies comprise both autosomal recessive and autosomal dominant muscle disorders that have a variable expression (phenotype) in individuals, but all share similar structural features in the muscles.
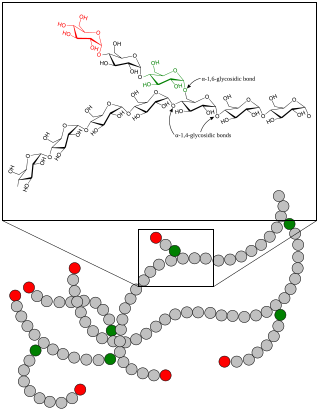
Glycogen storage disease type IV (GSD IV), or Andersen's Disease, is a form of glycogen storage disease, which is caused by an inborn error of metabolism. It is the result of a mutation in the GBE1 gene, which causes a defect in the glycogen branching enzyme. Therefore, glycogen is not made properly and abnormal glycogen molecules accumulate in cells; most severely in cardiac and muscle cells. The severity of this disease varies on the amount of enzyme produced. GSD IV is autosomal recessive, which means each parent has a mutant copy of the gene, but show no symptoms of the disease. Having an autosomal recessive inheritance pattern, males and females are equally likely to be affected by Andersen's disease. Classic Andersen's disease typically becomes apparent during the first few months after the patient is born. Approximately 1 in 20,000 to 25,000 newborns have a glycogen storage disease. Andersen's disease affects 1 in 800,000 individuals worldwide, with 3% of all GSDs being type IV. The disease was described and studied first by Dorothy Hansine Andersen.
Glycogen storage disease type III (GSD III) is an autosomal recessive metabolic disorder and inborn error of metabolism (specifically of carbohydrates) characterized by a deficiency in glycogen debranching enzymes. It is also known as Cori's disease in honor of the 1947 Nobel laureates Carl Cori and Gerty Cori. Other names include Forbes disease in honor of clinician Gilbert Burnett Forbes (1915–2003), an American physician who further described the features of the disorder, or limit dextrinosis, due to the limit dextrin-like structures in cytosol. Limit dextrin is the remaining polymer produced after hydrolysis of glycogen. Without glycogen debranching enzymes to further convert these branched glycogen polymers to glucose, limit dextrinosis abnormally accumulates in the cytoplasm.

Centronuclear myopathies (CNM) are a group of congenital myopathies where cell nuclei are abnormally located in the center of muscle cells instead of their normal location at the periphery.

Metabolic myopathies are myopathies that result from defects in biochemical metabolism that primarily affect muscle. They are generally genetic defects that interfere with the ability to create energy, causing a low ATP reservoir within the muscle cell.

Neutral lipid storage disease is a congenital autosomal recessive disorder characterized by accumulation of triglycerides in the cytoplasm of leukocytes, muscle, liver, fibroblasts, and other tissues. It commonly occurs as one of two subtypes, cardiomyopathic neutral lipid storage disease (NLSD-M), or ichthyotic neutral lipid storage disease (NLSD-I) which is also known as Chanarin–Dorfman syndrome), which are characterized primarily by myopathy and ichthyosis, respectively. Normally, the ichthyosis that is present is typically non-bullous congenital ichthyosiform erythroderma which appears as white scaling.
Hoffmann syndrome is a rare form of hypothyroid myopathy and is not to be confused with Werdnig-Hoffmann disease.
Acquired non-inflammatory myopathy (ANIM) is a neuromuscular disorder primarily affecting skeletal muscle, most commonly in the limbs of humans, resulting in a weakness or dysfunction in the muscle. A myopathy refers to a problem or abnormality with the myofibrils, which compose muscle tissue. In general, non-inflammatory myopathies are a grouping of muscular diseases not induced by an autoimmune-mediated inflammatory pathway. These muscular diseases usually arise from a pathology within the muscle tissue itself rather than the nerves innervating that tissue. ANIM has a wide spectrum of causes which include drugs and toxins, nutritional imbalances, acquired metabolic dysfunctions such as an acquired defect in protein structure, and infections.

X-linked myopathy with excessive autophagy (XMEA) is a rare childhood onset disease characterized by slow progressive vacuolation and atrophy of skeletal muscle. There is no known cardiac or intellectual involvement.

Adult polyglucosan body disease (APBD) is a rare monogenic glycogen storage disorder caused by an inborn error of metabolism. Symptoms can emerge any time after the age of 30. Early symptoms include trouble controlling urination, trouble walking, and lack of sensation in the legs. People eventually develop dementia.
Autophagic vacuolar myopathy (AVM) consists of multiple rare genetic disorders with common histological and pathological features on muscle biopsy. The features highlighted are vacuolar membranes of the autophagic vacuoles having sarcolemmal characteristics and an excess of autophagic vacuoles. There are currently five types of AVM identified. The signs and symptoms become more severe over the course of the disease. It begins with an inability to pick up small objects and progresses to difficulty in walking. The age of onset varies from early childhood to late adulthood, affecting people of all ages.