Leigh syndrome is an inherited neurometabolic disorder that affects the central nervous system. It is named after Archibald Denis Leigh, a British neuropsychiatrist who first described the condition in 1951. Normal levels of thiamine, thiamine monophosphate, and thiamine diphosphate are commonly found, but there is a reduced or absent level of thiamine triphosphate. This is thought to be caused by a blockage in the enzyme thiamine-diphosphate kinase, and therefore treatment in some patients would be to take thiamine triphosphate daily. While the majority of patients typically exhibit symptoms between the ages of 3 and 12 months, instances of adult onset have also been documented.
Immunodeficiency, also known as immunocompromisation, is a state in which the immune system's ability to fight infectious diseases and cancer is compromised or entirely absent. Most cases are acquired ("secondary") due to extrinsic factors that affect the patient's immune system. Examples of these extrinsic factors include HIV infection and environmental factors, such as nutrition. Immunocompromisation may also be due to genetic diseases/flaws such as SCID.

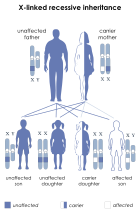
X-linked recessive inheritance is a mode of inheritance in which a mutation in a gene on the X chromosome causes the phenotype to be always expressed in males and in females who are homozygous for the gene mutation, see zygosity. Females with one copy of the mutated gene are carriers.

Neisseria meningitidis, often referred to as the meningococcus, is a Gram-negative bacterium that can cause meningitis and other forms of meningococcal disease such as meningococcemia, a life-threatening sepsis. The bacterium is referred to as a coccus because it is round, and more specifically a diplococcus because of its tendency to form pairs.

X-linked agammaglobulinemia (XLA) is a rare genetic disorder discovered in 1952 that affects the body's ability to fight infection. As the form of agammaglobulinemia that is X-linked, it is much more common in males. In people with XLA, the white blood cell formation process does not generate mature B cells, which manifests as a complete or near-complete lack of proteins called gamma globulins, including antibodies, in their bloodstream. B cells are part of the immune system and normally manufacture antibodies, which defend the body from infections by sustaining a humoral immunity response. Patients with untreated XLA are prone to develop serious and even fatal infections. A mutation occurs at the Bruton's tyrosine kinase (Btk) gene that leads to a severe block in B cell development and a reduced immunoglobulin production in the serum. Btk is particularly responsible for mediating B cell development and maturation through a signaling effect on the B cell receptor BCR. Patients typically present in early childhood with recurrent infections, in particular with extracellular, encapsulated bacteria. XLA is deemed to have a relatively low incidence of disease, with an occurrence rate of approximately 1 in 200,000 live births and a frequency of about 1 in 100,000 male newborns. It has no ethnic predisposition. XLA is treated by infusion of human antibody. Treatment with pooled gamma globulin cannot restore a functional population of B cells, but it is sufficient to reduce the severity and number of infections due to the passive immunity granted by the exogenous antibodies.
Hypogammaglobulinemia is an immune system disorder in which not enough gamma globulins are produced in the blood. This results in a lower antibody count, which impairs the immune system, increasing risk of infection. Hypogammaglobulinemia may result from a variety of primary genetic immune system defects, such as common variable immunodeficiency, or it may be caused by secondary effects such as medication, blood cancer, or poor nutrition, or loss of gamma globulins in urine, as in nonselective glomerular proteinuria. Patients with hypogammaglobulinemia have reduced immune function; important considerations include avoiding use of live vaccines, and take precautionary measures when traveling to regions with endemic disease or poor sanitation such as receiving immunizations, taking antibiotics abroad, drinking only safe or boiled water, arranging appropriate medical cover in advance of travel, and ensuring continuation of any immunoglobulin infusions needed.
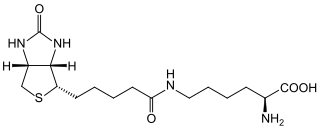
Biotinidase deficiency is an autosomal recessive metabolic disorder in which biotin is not released from proteins in the diet during digestion or from normal protein turnover in the cell. This situation results in biotin deficiency.

Meningococcal disease describes infections caused by the bacterium Neisseria meningitidis. It has a high mortality rate if untreated but is vaccine-preventable. While best known as a cause of meningitis, it can also result in sepsis, which is an even more damaging and dangerous condition. Meningitis and meningococcemia are major causes of illness, death, and disability in both developed and under-developed countries.

Properdin is a protein that in humans is encoded by the CFP gene.

Alström syndrome (AS), also called Alström–Hallgren syndrome, is a very rare autosomal recessive genetic disorder characterised by childhood obesity and multiple organ dysfunction. Symptoms include early-onset type 2 diabetes, cone-rod dystrophy resulting in blindness, sensorineural hearing loss and dilated cardiomyopathy. Endocrine disorders typically also occur, such as hypergonadotrophic hypogonadism and hypothyroidism, as well as acanthosis nigricans resulting from hyperinsulinemia. Developmental delay is seen in almost half of people with Alström syndrome.
The Kidd antigen system are proteins found in the Kidd's blood group, which act as antigens, i.e., they have the ability to produce antibodies under certain circumstances. The Jk antigen is found on a protein responsible for urea transport in the red blood cells and the kidney. They are important in transfusion medicine. People with two Jk(a) antigens, for instance, may form antibodies against donated blood containing two Jk(b) antigens. This can lead to hemolytic anemia, in which the body destroys the transfused blood, leading to low red blood cell counts. Another disease associated with the Jk antigen is hemolytic disease of the newborn, in which a pregnant woman's body creates antibodies against the blood of her fetus, leading to destruction of the fetal blood cells. Hemolytic disease of the newborn associated with Jk antibodies is typically mild, though fatal cases have been reported.

Complement C2 is a protein that in humans is encoded by the C2 gene. The protein encoded by this gene is part of the classical pathway of the complement system, acting as a multi-domain serine protease. Deficiency of C2 has been associated with certain autoimmune diseases.
Mannose-binding lectin (MBL), also called mannan-binding lectin or mannan-binding protein (MBP), is a lectin that is instrumental in innate immunity as an opsonin and via the lectin pathway.
George D. Heist (1886–1920) was an immunologist specializing in the study of infections of meningococcal bacteria that often result in meningococcal disease, which is well known as highly lethal and debilitating, and extremely difficult to treat.

Complement deficiency is an immunodeficiency of absent or suboptimal functioning of one of the complement system proteins. Because of redundancies in the immune system, many complement disorders are never diagnosed. Some studies estimate that less than 10% are identified. Hypocomplementemia may be used more generally to refer to decreased complement levels, while secondary complement disorder means decreased complement levels that are not directly due to a genetic cause but secondary to another medical condition.
NmVac4-A/C/Y/W-135 is the commercial name of the polysaccharide vaccine against the bacterium that causes meningococcal meningitis. The product, by JN-International Medical Corporation, is designed and formulated to be used in developing countries for protecting populations during meningitis disease epidemics.

Complement 3 deficiency is a genetic condition affecting complement component 3 (C3). People can suffer from either primary or secondary C3 deficiency. Primary C3 deficiency refers to an inherited autosomal-recessive disorder that involves mutations in the gene for C3. Secondary C3 deficiency results from a lack of factor I or factor H, two proteins that are key for the regulation of C3. Both primary and secondary C3 deficiency are characterized by low levels or absence of C3.
Surfactant metabolism dysfunction is a condition where pulmonary surfactant is insufficient for adequate respiration. Surface tension at the liquid-air interphase in the alveoli makes the air sacs prone to collapsing post expiration. This is due to the fact that water molecules in the liquid-air surface of alveoli are more attracted to one another than they are to molecules in the air. For sphere-like structures like alveoli, water molecules line the inner walls of the air sacs and stick tightly together through hydrogen bonds. These intermolecular forces put great restraint on the inner walls of the air sac, tighten the surface all together, and unyielding to stretch for inhalation. Thus, without something to alleviate this surface tension, alveoli can collapse and cannot be filled up again. Surfactant is essential mixture that is released into the air-facing surface of inner walls of air sacs to lessen the strength of surface tension. This mixture inserts itself among water molecules and breaks up hydrogen bonds that hold the tension. Multiple lung diseases, like ISD or RDS, in newborns and late-onsets cases have been linked to dysfunction of surfactant metabolism.
BENTA disease is a rare genetic disorder of the immune system. BENTA stands for "B cell expansion with NF-κB and T cell anergy" and is caused by germline heterozygous gain-of-function mutations in the gene CARD11. This disorder is characterized by polyclonal B cell lymphocytosis with onset in infancy, splenomegaly, lymphadenopathy, mild immunodeficiency, and increased risk of lymphoma. Investigators Andrew L. Snow and Michael J. Lenardo at the National Institute of Allergy and Infectious Diseases at the U.S. National Institutes of Health first characterized BENTA disease in 2012. Dr. Snow's current laboratory at the Uniformed Services University of the Health Sciences is now actively studying this disorder.
PGM3 deficiency is a rare genetic disorder of the immune system associated with diminished phosphoglucomutase 3 function. PGM3 is an enzyme which in humans is encoded by gene PGM3. This disorder manifests as severe atopy, immune deficiency, autoimmunity, intellectual disability, and hypomyelination. In 2014, Investigators Atfa Sassi at the Pasteur Institute of Tunis, Sandra Lazaroski at the University Medical Center Freiburg, and Gang Wu at the Imperial College London, identified PGM3 mutations in nine patients from four consanguineous families. In the same year, a researchers from the laboratories of Joshua Milner and Helen Su at the National Institute of Allergy and Infectious Disease at the U.S. National Institutes of Health described PGM3 deficiency in eight additional patients from two families.