It is believed to be inherited in an X-linked recessive manner, which means a geneticmutation causing the disorder is located on the X chromosome, and while two copies of the mutated gene must be inherited for a female to be born with the disorder, just one copy is sufficient to cause a male to be born with the disorder. Nasodigitoacoustic syndrome is likely caused by a mutated gene located on the X chromosome between positions Xq22.2–q28.
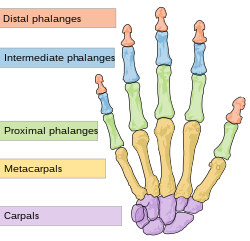
The distal, or terminal phalanges (orange) are at the end of the fingers and toes.
Nasodigitoacoustic syndrome is congenital and is characterized by a number of nasal, facial and cranial features. These include a broad and high, sometimes depressed nasal bridge (top of the nose, between the eyes) and a flattened nasal tip.[2][6][7] This can give the nose a shortened, arch-like appearance.[8]Hypertelorism (unusually wide-set eyes),[4] prominent frontal bones and supraorbital ridge (the eyebrow ridge), bilateral epicanthic folds (an extra flap of skin over the eyelids), a broad forehead and an overall enlarged head circumference have also been observed. A bulging of the upper lip with an exaggerated cupid's bow shape,[9] and maxillary hypoplasia (underdevelopment of the upper jaw) with retraction have also been reported.[2][7][10]
Several anomalies affecting the digits (fingers and toes) have been observed with the syndrome. A broadening of the thumbs and big toes (halluces) was reported in two brothers. The broadening was apparent in all distal phalanges of the fingers, although the pinkies were unaffected yet appeared to be clinodactylic (warped, or bent toward the other fingers).[2] Additional reports described this broadness of the thumbs and big toes, with brachydactyly (shortness) in the distal phalanges of the other digits except the pinkies in affected individuals. On X-rays of a two-year-old boy with the disorder, the brachydactyly was shown to be caused by shortening of epiphyses (joint-ends) of the distal phalanges.[7][11] The broadness and brachydactyly of the big toes in particular may give them a stunted, rounded and stub-like appearance.[8]
The auditory, or "acoustic" abnormalities observed with the syndrome include sensorineural hearing loss and hoarseness. Two affected Turkish brothers with a mild form of this hearing loss, and a hoarse voice were reported. A laryngoscopic examination of both brothers revealed swelling of the vocal cords, and a malformed epiglottis.[6] Sensorineural-associated hearing impairment and hoarseness was also observed in a 10-year-old girl and her father,[10] and in a number of other cases.[3][7]
Schematic of the human X chromosomeNasodigitoacoustic syndrome has an X-linked recessive pattern of inheritance.
Nasodigitoacoustic syndrome is thought to be caused by a mutation in a gene on the X chromosome. A 2007 study concluded, based on analysis of microsatellite markers (small gene sequences found in common among individuals having the same ethnicity, ancestry or genetic disease) of the family described by Keipert, that this gene was likely located on the long arm of the X chromosome between positions Xq22.2–q28. This is not definitive, however, and no specific gene has been named.[3]
The syndrome is strongly believed to be inherited in an X-linked recessive manner.[3] When a female carries a mutated gene on one of her two copies of the X chromosome, there is a 50% chance of passing the mutation on to her children. Much like her, a daughter inheriting this mutation will be a carrier, but will not herself have the associated disease. However, a son who inherits the mutation will have the disease; this is because males have only one copy of the X chromosome and therefore could only express the disease mutation.
This form of inheritance for Nasodigitoacoustic syndrome is not yet absolute, though, as a girl has been reported with the disorder. It is suggested that further analysis is needed for the inheritance to be formally established.[7][10]
The mutations causing this syndrome have been mapped to the glypican 4 (GPC4) gene.[12] This gene is located on the long arm of chromosome X (Xq26.2).
Diagnosis
The constellation of anomalies seen with nasodigitoacoustic syndrome result in a distinct diagnosis. The diagnostic criteria for the disorder are broad distal phalanges of the thumbs and big toes, accompanied by a broad and shortened nose, sensorineural hearing loss and developmental delay, with predominant occurrence in males.[4][9]
Classification
Nasodigitoacoustic syndrome is similar to several syndromes that share its features.[4][7] Brachydactyly of the distal phalanges, sensorineural deafness and pulmonary stenosis are common with Keutel syndrome.[13] In Muenke syndrome, developmental delay, distal brachydactyly and sensorineural hearing loss are reported; features of Teunissen-Cremers syndrome include nasal aberrations and broadness of the thumbs and big toes, also with brachydactyly.[14][15] Broad thumbs and big toes are primary characteristics of Rubinstein syndrome.[16]
Management
A number of features found with nasodigitoacoustic syndrome can be managed or treated. Sensorineural hearing loss in humans may be caused by a loss of hair cells (sensory receptors in the inner ear that are associated with hearing). This can be hereditary and/or within a syndrome, as is the case with nasodigitoacoustic syndrome,[4] or attributed to infections such as viruses. For the management of sensorineural hearing loss, hearing aids have been used. Treatments, depending upon the cause and severity, may include a pharmacological approach (i.e., the use of certain steroids), or surgical intervention, like a cochlear implant.[17][18][19]
Pulmonary, or pulmonic stenosis is an often congenital narrowing of the pulmonary valve; it can be present in nasodigitoacoustic-affected infants.[4] Treatment of this cardiac abnormality can require surgery, or non-surgical procedures like balloon valvuloplasty (widening the valve with a balloon catheter).[20]
History
The syndrome was initially described in 1973 by James A. Keipert and associates. They reported of two brothers with broad distal phalanges, sensorineural hearing loss, and facial features consistent with what would become known as Keipert or "nasodigitoacoustic" syndrome.[2][4]
Epidemiology
Although no specific rate of incidence has been determined, the syndrome is considered a rare disease by both the Office of Rare Diseases (ORDR) at the National Institutes of Health, and Orphanet. This suggests, respectively, that nasodigitoacoustic syndrome affects less than 200,000 people in the U.S., or affects no greater than 1 per 2,000 people in Europe.[5]
1 2 3 4 5 Amor, D. J.; Dahl, H-H.; Bahlo, M.; Bankier, A. (Oct 2007). "Keipert syndrome (Nasodigitoacoustic syndrome) is X-linked and maps to Xq22.2–Xq28". American Journal of Medical Genetics Part A. 143A (19): 2236–2241. doi:10.1002/ajmg.a.31917. PMID17726694. S2CID34320632.
1 2 3 4 5 6 Nik-Zainal, S.; Holder, S. E.; Cruwys, M.; Hall, C. M.; Shaw-Smith, C. (Jul 2008). "Keipert syndrome: two further cases and review of the literature". Clinical Dysmorphology. 17 (3): 169–175. doi:10.1097/MCD.0b013e3282f4afc3. PMID18541962.
1 2 3 Dumic, M.; Kokic, D. D.; Matic, T.; Potocki, K. (Nov 2006). "Daughter and her mildly affected father with Keipert syndrome". American Journal of Medical Genetics Part A. 140A (22): 2488–2492. doi:10.1002/ajmg.a.31489. PMID17036315. S2CID2978286.
↑ Reardon, W.; Hall, C. M. (Apr 2003). "Broad thumbs and halluces with deafness: A patient with Keipert syndrome". American Journal of Medical Genetics. 118A (1): 86–89. doi:10.1002/ajmg.a.10063. PMID12605449. S2CID27419345.
↑ Amor DJ, Stephenson SEM, Mustapha M, Mensah MA, Ockeloen CW, Lee WS, Tankard RM, Phelan DG, Shinawi M, de Brouwer APM, Pfundt R, Dowling C, Toler TL, Sutton VR, Agolini E, Rinelli M, Capolino R, Martinelli D, Zampino G, Dumić M, Reardon W, Shaw-Smith C, Leventer RJ, Delatycki MB, Kleefstra T7, Mundlos S, Mortier G, Bahlo M, Allen NJ, Lockhart P (2019) Pathogenic variants in GPC4 cause Keipert syndrome. Am J Hum Genet
↑ Kikidis, D.; Nikolopoulos, T. P.; Kampessis, G.; Stamatiou, G.; Chrysovergis, A. (2011). "Sudden Sensorineural Hearing Loss: Subclinical Viral and Toxoplasmosis Infections as Aetiology and How They Alter the Clinical Course". ORL. 73 (2): 110–115. doi:10.1159/000324210. PMID21389742. S2CID25767318.
↑ Ali Khan, M.; Al-Yousef, S.; Huhta, J.; Bricker, J.; Mullins, C.; Sawyer, W. (May 1989). "Critical pulmonary valve stenosis in patients less than 1 year of age: Treatment with percutaneous gradational balloon pulmonary valvuloplasty". American Heart Journal. 117 (5): 1008–1014. doi:10.1016/0002-8703(89)90854-5. PMID2711961.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.