
Fraser syndrome is an autosomal recessive congenital disorder, identified by several developmental anomalies. Fraser syndrome is named for the geneticist George R. Fraser, who first described the syndrome in 1962.

Behr syndrome is characterized by the association of early-onset optic atrophy with spinocerebellar degeneration resulting in ataxia, pyramidal signs, peripheral neuropathy and developmental delay.
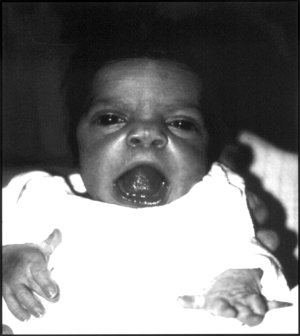
Robinow syndrome is an extremely rare genetic disorder characterized by short-limbed dwarfism, abnormalities in the head, face, and external genitalia, as well as vertebral segmentation. The disorder was first described in 1969 by human geneticist Meinhard Robinow, along with physicians Frederic N. Silverman and Hugo D. Smith, in the American Journal of Diseases of Children. By 2002, over 100 cases had been documented and introduced into medical literature.

Larsen syndrome (LS) is a congenital disorder discovered in 1950 by Larsen and associates when they observed dislocation of the large joints and face anomalies in six of their patients. Patients with Larsen syndrome normally present with a variety of symptoms, including congenital anterior dislocation of the knees, dislocation of the hips and elbows, flattened facial appearance, prominent foreheads, and depressed nasal bridges. Larsen syndrome can also cause a variety of cardiovascular and orthopedic abnormalities. This rare disorder is caused by a genetic defect in the gene encoding filamin B, a cytoplasmic protein that is important in regulating the structure and activity of the cytoskeleton. The gene that influences the emergence of Larsen syndrome is found in chromosome region, 3p21.1-14.1, a region containing human type VII collagen gene. Larsen syndrome has recently been described as a mesenchyme disorder that affects the connective tissue of an individual. Autosomal dominant and recessive forms of the disorder have been reported, although most cases are autosomal dominant. Reports have found that in Western societies, Larsen syndrome can be found in one in every 100,000 births, but this is most likely an underestimate because the disorder is frequently unrecognized or misdiagnosed.
An osteochondrodysplasia, or skeletal dysplasia, is a disorder of the development of bone and cartilage. Osteochondrodysplasias are rare diseases. About 1 in 5,000 babies are born with some type of skeletal dysplasia. Nonetheless, if taken collectively, genetic skeletal dysplasias or osteochondrodysplasias comprise a recognizable group of genetically determined disorders with generalized skeletal affection. These disorders lead to disproportionate short stature and bone abnormalities, particularly in the arms, legs, and spine. Skeletal dysplasia can result in marked functional limitation and even mortality.

Micrognathism is a condition where the jaw is undersized. It is also sometimes called mandibular hypoplasia. It is common in infants, but is usually self-corrected during growth, due to the jaws' increasing in size. It may be a cause of abnormal tooth alignment and in severe cases can hamper feeding. It can also, both in adults and children, make intubation difficult, either during anesthesia or in emergency situations.

Multiple epiphyseal dysplasia (MED), also known as Fairbank's disease, is a rare genetic disorder that affects the growing ends of bones. Long bones normally elongate by expansion of cartilage in the growth plate near their ends. As it expands outward from the growth plate, the cartilage mineralizes and hardens to become bone (ossification). In MED, this process is defective.

Pseudoachondroplasia is an inherited disorder of bone growth. It is a genetic autosomal dominant disorder. It is generally not discovered until 2–3 years of age, since growth is normal at first. Pseudoachondroplasia is usually first detected by a drop of linear growth in contrast to peers, a waddling gait or arising lower limb deformities.

Matrix Gla protein (MGP) is member of a family of vitamin K2 dependent, Gla-containing proteins. MGP has a high affinity binding to calcium ions, similar to other Gla-containing proteins. The protein acts as an inhibitor of vascular mineralization and plays a role in bone organization.
Williams–Campbell syndrome (WCS) is a disease of the airways where cartilage in the bronchi is defective. It is a form of congenital cystic bronchiectasis. This leads to collapse of the airways and bronchiectasis. It acts as one of the differential to allergic bronchopulmonary aspergillosis. WCS is a deficiency of the bronchial cartilage distally.
Antley–Bixler syndrome is a rare, severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.

Fibrochondrogenesis is a rare autosomal recessive form of osteochondrodysplasia, causing abnormal fibrous development of cartilage and related tissues.
Gerodermia osteodysplastica (GO) is a rare autosomal recessive connective tissue disorder included in the spectrum of cutis laxa syndromes.
X-linked recessive chondrodysplasia punctata is a type of chondrodysplasia punctata that can involve the skin, hair, and cause short stature with skeletal abnormalities, cataracts, and deafness.
Congenital hypoplastic anemia is a congenital disorder that occasionally also includes leukopenia and thrombocytopenia and is characterized by deficiencies of red cell precursors.

Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
Fryns syndrome is an autosomal recessive multiple congenital anomaly syndrome that is usually lethal in the neonatal period. Fryns (1987) reviewed the syndrome.

Amelogenesis imperfecta (AI) is a congenital disorder which presents with a rare abnormal formation of the enamel or external layer of the crown of teeth, unrelated to any systemic or generalized conditions. Enamel is composed mostly of mineral, that is formed and regulated by the proteins in it. Amelogenesis imperfecta is due to the malfunction of the proteins in the enamel as a result of abnormal enamel formation via amelogenesis.
Opsismodysplasia is a type of skeletal dysplasia first described by Zonana and associates in 1977, and designated under its current name by Maroteaux (1984). Derived from the Greek opsismos ("late"), the name "opsismodysplasia" describes a delay in bone maturation. In addition to this delay, the disorder is characterized by micromelia, particularly of the hands and feet, delay of ossification, platyspondyly, irregular metaphyses, an array of facial aberrations and respiratory distress related to chronic infection. Opsismodysplasia is congenital, being apparent at birth. It has a variable mortality, with some affected individuals living to adulthood. The disorder is rare, with an incidence of less than 1 per 1,000,000 worldwide. It is inherited in an autosomal recessive pattern, which means the defective (mutated) gene that causes the disorder is located on an autosome, and the disorder occurs when two copies of this defective gene are inherited. No specific gene has been found to be associated with the disorder. It is similar to spondylometaphyseal dysplasia, Sedaghatian type.
