Neurofibromatosis type I (NF-1), or von Recklinghausen syndrome, is a complex multi-system neurocutaneous disorder caused by a subset of genetic mutations at the neurofibromin 1 (NF1) locus. Other conditions associated with mutation of the NF1 gene include Watson syndrome.[2]NF1 is a gene on chromosome 17 that is responsible for production of a protein (neurofibromin) which is needed for normal function in many human cell types. NF-1 causes tumors along the nervous system that can grow anywhere on the body. NF-1 is one of the most common genetic disorders and is not limited to any person's race or sex. NF-1 is an autosomal dominant disorder, which means that mutation or deletion of one copy (or allele) of the NF-1 gene is sufficient for the development of NF-1, although presentation varies widely and is often different even between relatives affected by NF-1.[3]
As of 2015[update], there are at least 100,000 people in the U.S. and about 25,000 people in the UK who have been diagnosed with NF. Common symptoms of NF-1 include brownish-red spots in the colored part of the eye called Lisch nodules, benign skin tumors called neurofibromas, and larger benign tumors of nerves called plexiform neurofibromas, scoliosis (curvature of the spine), learning disabilities, vision disorders, mental disabilities, multiple café au lait spots and epilepsy. While some people have major complications, others with the condition can lead productive and full lives.
NF-1 is a developmental syndrome caused by germline mutations in neurofibromin, a gene that is involved in the RAS pathway (RASopathy). Due to its rarity, and to the fact that genetic diagnosis has been used only in recent years, in the past NF-1 was in some cases confused with Legius syndrome, another syndrome with vaguely similar symptoms, including cafe au lait spots.[4]
NF-1 is an age-specific disease; most signs of NF-1 are visible after birth (during infancy), but many symptoms of NF-1 occur as the person ages and has hormonal changes. NF-1 was formerly known as von Recklinghausen disease, after the researcher who first documented the disorder, Friedrich Daniel von Recklinghausen.[5]
The severity of NF-1 varies widely, and little is known about what causes a person to have more severe or less severe symptoms. Even within the same family (as there is a 50% chance that a parent will pass their condition to their offspring), levels of severity can vary enormously.[3] 60% of people with NF-1 have mild cases, with few symptoms that have very little effect in their day-to-day lives. About 20% of people with NF-1 have what are considered moderate cases, with several symptoms that usually have a few cosmetic effects. The other 20% have severe cases, with several symptoms that affect the person's quality of life. Even in this last group, symptoms are rarely life-threatening.[6]
Signs and symptoms
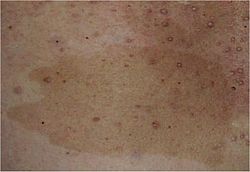
Café au lait spot characteristic of NF1Diagnostic criteria of neurofibromatosis type I, requiring at least 2 of the mentioned items.
The following is a list of conditions and complications associated with NF-1, and, where available, age range of onset and progressive development, occurrence percentage of NF-1 population, method of earliest diagnosis, and treatments and related medical specialties.[8][9] The progression of the condition is roughly as follows:
Congenital musculoskeletal disorders may or may not be present.
Cutaneous conditions may be observed in early infancy.
Small tumors may arise in the retina which can eventually lead to blindness. Also, Lisch nodules may grow on the iris, but these are harmless.
Learning disabilities may arise in preschool children.
Neurofibromas may occur and can sometimes cause many dependent neurological conditions and cutaneous and skeletal disfigurement.
Depression and social anxiety may occur as a result of disabilities caused by the condition.
Neurofibromas may, in 8-13% of cases, transition into cancer, which can be fatal.[10]
In NF-1, there can be a generalized abnormality of the soft tissues in the fetus, which is referred to as mesodermaldysplasia, resulting in maldevelopment of skeletal structures.
Meningoceles and formation of cystic diverticula of the dura of the spine, unrelated to Spina bifida
Radiographically, dural ectasia can lead to scalloping of the posterior vertebral bodies and to the formation of cystic diverticula of the dura of the spine. This may result in temporary or permanent loss of lower extremity sensorimotor function.[11]
Focal scoliosis and/or kyphosis are the most common skeletal manifestation of NF-1, occurring in 20% of affected patients. Approximately 25% of patients will require corrective surgery.
Skeletal muscle weakness and motor control deficits
Deficits in motor function in NF-1 have been long recognised and have been historically attributed to nerve dysfunction. In recent years however, studies suggest NF-1 is associated with a primary problem in muscle function (myopathy).[12]
Clinical findings in people with NF-1 include:
Reduced skeletal muscle size
Reduced exercise capacity
Muscle weakness (the most recent study reports between 30–50% reduced upper and lower limb muscle strength in NF-1 children compare with matched controls[13]).
Studies in genetically modified mice have thus far confirmed that the NF1 gene is vital for normal muscle development and metabolism. Knockout of the NF1 gene in muscle results in deregulated lipid metabolism and muscle weakness.[12][14]
NF-1 is a disease in the RASopathy family of diseases, which include Costello syndrome, Noonan syndrome, and cardiofaciocutaneous syndrome. The RASopathies also present with skeletal muscle weakness.[15] It is likely that impaired muscle function in these disorders is linked to altered Ras/MAPK signalling, however, the precise molecular mechanisms remain unknown.[12]
Malformation of the facial bones or of the eye sockets (lambdoid suture defects, sphenoid dysplasia)
Unilateral overgrowth of a limb. When a plexiform neurofibroma manifests on a leg or arm, it will cause extra blood circulation, and may thus accelerate the growth of the limb. This may cause considerable difference in length between left and right limbs. To equalize the difference during childhood, there is an orthopedic surgery called epiphysiodesis, where growth at the epiphyseal (growth) plate is halted. It can be performed on one side of the bone to help correct an angular deformity, or on both sides to stop growth of that bone completely. The surgery must also be carefully planned with regard to timing, as it is non-reversible. The goal is that the limbs are at near-equal length at end of growth.
Flat pigmented lesions of the skin called café au lait spots are hyperpigmented lesions that may vary in color from light brown to dark brown; this is reflected by the name of the condition, which means "coffee with milk". The borders may be smooth or irregular. These spots can grow from birth and can continue to grow throughout the person's lifetime. They can increase in size and numbers during puberty and during pregnancies. They are present in about 99% of patients of European origin and in about 93% of patients of Indian origin.[16]
Dermal neurofibroma, manifested as single or multiple firm, rubbery bumps of varying sizes on a person's skin. Age of onset is puberty. Progressive in number and size. Not malignant. Can be treated with CO2 lasers or by removal by a plastic surgeon specialized in NF1.[17][18]
Optic nerve gliomas along one or both optic nerves or the optic chiasm can cause bulging of the eyes, involuntary eye movement, squinting, and / or vision loss. Treatment may include surgery, radiation +/- steroids, or chemotherapy (in children).[19]
Neurobehavioral developmental disorder
The most common complication in patients with NF-1 is cognitive and learning disability. These cognitive problems have been shown to be present in approximately 90% of children and adults with NF-1 and have significant effects on their schooling and everyday life.[20] These cognitive problems have been shown to be stable into adulthood mainly in the mid 20s to early 30s and do not get worse unlike some of the other physical symptoms of NF-1.[21] The most common cognitive problems are with perception, executive functioning and attention. Disorders include:
Approximately 42% of children with NF-1 have symptoms of autism, with 36.78% of them being severe cases, 33.33% being mild to moderate cases, and 29.89% of them having both symptoms of autism and ADHD.[22]
The primary neurologic involvement in NF-1 is of the peripheral nervous system, and secondarily of the central nervous system. Schwannomatosis is a rare condition defined by the presence of multiple benign tumors of nerves that are frequently very painful. In addition to pain, weakness is a common problem. Symptoms usually begin in young or mid-adult years.[citation needed]
Peripheral neuropathy
Neurofibroma
A neurofibroma is a lesion of the peripheral nervous system. Its cellular lineage is uncertain, and may derive from Schwann cells, other perineural cell lines, or fibroblasts. Neurofibromas may arise sporadically, or in association with NF-1.
Neurofibroma conditions are progressive and include:
Plexiform neurofibroma: Often congenital. Lesions are composed of sheets of neurofibromatous tissue that may infiltrate and encase major nerves, blood vessels, and other vital structures. These lesions are difficult and sometimes impossible to routinely resect without causing any significant damage to surrounding nerves and tissue.
Schwannomas, peripheral nerve-sheath tumors which are seen with increased frequency in NF-1. The major distinction between a schwannoma and a solitary neurofibroma is that a schwannoma can be resected while sparing the underlying nerve, whereas resection of a neurofibroma requires the sacrifice of the underlying nerve.
Nerve root neurofibroma.
Bones, especially the ribs, can develop chronic erosions (pits) from the constant pressure of adjacent neurofibroma or schwannoma. Similarly, the neural foramen of the spine can be widened due to the presence of a nerve root neurofibroma or schwannoma. Surgery may be needed when NF-1 related tumors compress organs or other structures.
Nerve sheath tumor
MRI image showing malignant peripheral nerve sheath tumor in the left tibia in neurofibromatosis type-1
Intracranially, NF-1 patients have a predisposition to develop glial tumors of the central nervous system, primarily optic nerve gliomas and associated blindness.[26]
Focally degenerative myelin
Another CNS manifestation of NF-1 is the so-called "unidentified bright object" or UBO, which is a lesion which has increased signal on a T2 weighted sequence of a magnetic resonance imaging examination of the brain. These UBOs are typically found in the Cerebral peduncle, pons, midbrain, globus pallidus, thalamus, and optic radiations. Their exact identity remains a bit of a mystery since they disappear over time (usually, by age 16), and they are not typically biopsied or resected. They may represent a focally degenerative bit of myelin.[citation needed]
Within the CNS, NF-1 manifests as a weakness of the dura, which is the tough covering of the brain and spine. Weakness of the dura leads to focal enlargement due to chronic exposure to the pressures of CSF pulsation, and typically presents as paraesthesia or loss of motor or sensory function.[11] It has been shown that dural ectasia occur near plexiform neurofibromas which may be infiltrative leading to weakening of the dura.[27]
Acetazolamide has shown promise as a treatment for this condition, and in very few cases do dural ectasia require surgery.[27]
Mental health
People with NF1 are at increased risk for experiencing social and emotional difficulties such as anxiety, depression, low self-esteem and/or body image, social withdrawal, difficulty forming interpersonal relationships, behavioural problems, and difficulties in school.[28] People with NF1 are much more likely to experience suicidal thoughts than the general population. One study found that 45% of people with NF had suicidal thoughts compared to 10% of a healthy control group.[29] Another study found that 46.5% were of people with NF1 were found to have at least one psychiatric comorbid diagnosis.[30]
Neurodivergence
Children and adults with NF-1 often have Autism and/or ADHD.
30 - 50% of people with NF1 also meet the diagnostic criteria for ADHD[31]
Children diagnosed with NF-1 may experience delayed or precocious puberty. Recent studies have correlated precocious puberty in individuals with NF-1 with the presence of optic pathway tumours.[33] Furthermore, the heights of children affected by NF-1 have been shown to increase normally until puberty, after which increases in height lessen when compared to healthy counterparts.[33] This eventually causes a shorter stature than expected in individuals with NF-1.
Mortality. Malignant nerve sheath tumor was the main cause of death (60%) in a study of 1895 patients with NF-1 from France in the time period 1980–2006 indicated excess mortality in NF-1 patients compared to the general population.[36] The cause of death was available for 58 (86.6%) patients. The study found excess mortality occurred among patients aged 10 to 40 years. Significant excess mortality was found in both males and females.
Breast cancer
Biological females with NF also have a five-fold increased risk of breast cancer and may have an increased breast cancer related mortality. The median survival for breast cancer in people with NF was 5 years vs. the reported median survival of over 20 years in the general population using the SEER database.[37][38]
In 1989, through linkage and cross over analyses, neurofibromin was localized to chromosome 17.[42] It was localized to the long arm of chromosome 17 by chance when researchers discovered chromosome exchanges between chromosome 17 with chromosome 1 and 22.[42] This exchange of genetic material presumably caused a mutation in the neurofibromin gene, leading to the NF1 phenotype. Two recurrent microdeletion types with microdeletion breakpoints located in paralogous regions flanking NF1 (proximal NF1-REP-a and distal NF1-REP-c for the 1.4 Mb type-1 microdeletion, and SUZ12 and SUZ12P for the 1.2 Mb type-2 microdeletion), are found in most cases.[43]
Structure
The neurofibromin gene was soon sequenced and found to be 350,000 base pairs in length.[44] However, the protein is 2818 amino acids long leading to the concept of splice variants.[45] For example, exon 9a, 23a and 48a are expressed in the neurons of the forebrain, muscle tissues and adult neurons respectively.[45]
Homology studies have shown that neurofibromin is 30% similar to proteins in the GTPase activating protein (GAP) family.[44] This homologous sequence is in the central portion of neurofibromin and being similar to the GAP family is recognized as a negative regulator of the Ras kinase.[46]
Additionally, being such a large protein, more active domains of the protein have been identified. One such domain interacts with the protein adenylyl cyclase,[47] and a second with collapsin response mediator protein.[48] Together, likely with domains yet to be discovered, neurofibromin regulates many of the pathways responsible for overactive cell proliferation, learning impairments, skeletal defects and plays a role in neuronal development.[49]
Inheritance and spontaneous mutation
NF-1 is inherited in an autosomal dominant fashion, although it can also arise due to spontaneous mutation.
The mutant gene is transmitted with an autosomal dominant pattern of inheritance, but up to 50% of NF-1 cases arise due to spontaneous mutation. The incidence of NF-1 is about 1 in 3500 live births.[50]
While the presence of NF-1 can be identified through prenatal testing the severity with which the condition will be expressed is impossible to determine.[52]
People with NF-1 have a 50% percent chance of passing the disorder to their offspring, but people can have a child born with NF-1 when they themselves do not have the condition. This is caused by a spontaneous mutation.
Post-natal testing
The National Institutes of Health (NIH) has created specific criteria for the diagnosis of NF-1. Two of these seven "Cardinal Clinical Features" are required for positive diagnosis.[53][54] There is practical flowchart to distinguish between NF1, NF2 and schwannomatosis.[55]
Six or more café-au-lait spots over 5mm in greatest diameter in pre-pubertal individuals and over 15mm in greatest diameter in post-pubertal individuals. Note that multiple café-au-lait spots alone are not a definitive diagnosis of NF-1 as these spots can be caused by a number of other conditions.
Two or more neurofibromas of any type or 1 plexiform neurofibroma
Two or more Lisch nodules (pigmented iris hamartomas)
A distinctive osseous lesion such as sphenoid dysplasia, or thinning of the long bone cortex with or without pseudarthrosis.
A first degree relative (parent, sibling, or offspring) with NF-1 by the above criteria.
Psychological and neurodevelopmental disorders
In addition to physical manifestations, patients with NF1 are at high risk of developing neurodevelopmental disorders, which result in learning difficulties, attention problems, and other behavioral or social challenges.[56] Studies have shown that children with NF1 are particularly prone to being affected by conditions such as Attention Deficit Hyperactivity Disorder (ADHD) or Autism Spectrum Disorder (ASD), as well as psychological disorders such as anxiety or depression, highlighting the importance of multidisciplinary evaluation and care for these patients.[56]
Autism spectrum disorder
A significant number of children with NF-1 exhibit symptoms commonly associated with Autism Spectrum Disorder (ASD), which can impact daily functioning.[57] These symptoms may include difficulties with flexibility and transitions, repetitive behaviors, challenges in social communication, social awareness, and adaptability.[57]
Some studies have identified subtle but significant differences between ASD symptomatology in individuals with NF-1 and those with idiopathic autism.[58] These differences include stronger eye contact, fewer repetitive behaviors, and more pronounced autistic mannerisms compared to non-syndromic ASD.[59] Enhanced language skills have also been noted in this population.[58] More than 90% of children with ASD + NF1 demonstrate clinically significant challenges in interpreting social signals and social communication during interactions.[58]
Discrepancies have been noted between parent-report questionnaires, such as the Social Responsiveness Scale (SRS), and clinical observation tools as in the case of the Autism Diagnostic Observation Schedule (ADOS), suggesting that restricted repetitive behaviors in NF-1 autism may be qualitatively different or less severe than in idiopathic autism, and therefore may go undetected in ADOS assessments.[58]
Studies indicate that parent-reported scores on the autistic mannerisms subscale of the SRS questionnaire were notably high, with two-thirds of children with NF-1 + ASD scoring in the severe problem range.[58]
Regarding items assessing imagination and creativity, children with NF-1 + ASD exhibit similar levels of impairment as the autism group, while being significantly more affected than children with only ASD.[58] Furthermore, no differences were observed between the groups on items measuring hyperactivity. Similarly, no evidence of group differences was found for the anxiety item, which is also associated with certain genetic disorders, such as Fragile X syndrome and Cornelia de Lange syndrome.[58]
Research suggests that Neurofibromatosis Type 1 and Tuberous Sclerosis (TSC) exhibit similarities in the symptomatology associated with Autism Spectrum Disorder.[59] These findings may indicate the existence of shared neurobiological characteristics between the two syndromes that influence the presentation of ASD symptoms.
Due to their fewer observed repetitive behaviors and improved eye contact, these children may not exhibit the typical characteristics of idiopathic autism in clinical settings, increasing the likelihood that they will be overlooked by clinicians.[58]
Unlike idiopathic ASD, both males and females seem to be equally affected, indicating a diminished protective effect of gender against ASD symptoms, akin to other syndromic causes of ASD.[56]
Other studies do not find a significant association between neurofibromatosis type 1 (NF1) and autism spectrum disorder (ASD).[60] While children with NF1 exhibit more autism-related symptoms and behavioral problems compared to typically developing children, these are often explained by the presence of ADHD and other concurrent issues, without reaching clinically significant levels.[60]
ADHD
Children with NF-1 may experience behavioral difficulties related to inattention, impulsivity, hyperactivity, and inflexibility.[57] Studies have shown that clinical criteria for diagnosing ADHD are met by 23% to 50% of children with NF-1.[61] Children with NF-1 may sometimes have attention difficulties without hyperactivity or behavioral problems. In such cases, attention deficits might go unnoticed without formal cognitive testing, although some children may have attention issues that, while not severe enough for an ADHD diagnosis, would still benefit from support.[57]
Individuals with neurofibromatosis type 1 often exhibit certain brain abnormalities known as T2 hyperintensities (visible on MRI scans), referred to as Unidentified Bright Objects (UBOs), which are located in specific brain regions such as the cerebellum, brainstem, thalamus, and basal ganglia—areas involved in motor signal processing and cognitive functions.[61] Some of these brain regions are also connected to attention-related networks, particularly those involved in cognitive flexibility and motor inhibition, which are essential for attention and behavior. It is well established that these networks are impaired in ADHD.[61]
Studies suggest that while ADHD symptoms may partially explain attention problems in NF-1, such as impulsivity, they do not fully account for other deficits like cognitive control.[61] Children with NF-1 often exhibit impairments in planning, spatial working memory, and response inhibition, independent of ADHD, suggesting that the impact of ADHD on their attention and executive functioning is limited.[61]
Although ADHD prevalence is a key factor in NF-1 cognition studies, comparisons between children with NF-1 and ADHD and those without ADHD have not consistently shown clear differences.[61] This creates confusion regarding how to differentiate the effects of NF-1 and ADHD on cognition. Moreover, focusing on ADHD symptoms might obscure attention issues that are specifically associated with NF-1.[61]
Results from tasks assigned in studies comparing NF-1 + ADHD groups with children who have only ADHD suggest that NF-1 + ADHD is not simply a combination of both conditions, nor does ADHD merely exacerbate attention problems in NF-1.[61] These differences are qualitative rather than quantitative, supporting the idea that some deficits are unique to NF-1 and independent of ADHD. Additionally, certain attention issues, such as intrinsic alertness and visual scanning among distractors, may be considered characteristic traits of NF-1.[61]
Treatment
Treatment for NF1 is limited, and there is currently no cure. Pain meds can be prescribed to help with pain. In some cases, growths may be removed surgically or reduced with radiation therapy. Treatment options are limited, given the tumours tendency to regrow following surgery and their propensity to transform into malignant tumours following radiation.[62] Additionally, surgery in these areas can cause further injury to nerves and additional neurological problems. The benefits of surgery should always be considered against its risks. Some NF tumours are inoperable.
The most common adverse reactions in adults include rash, diarrhea, nausea, musculoskeletal pain, vomiting, and fatigue.[65] The most common grade 3 or 4 laboratory abnormalities include increased creatine phosphokinase.[65] The most common adverse reactions in children include rash, diarrhea, musculoskeletal pain, abdominal pain, vomiting, headache, paronychia, left ventricular dysfunction, and nausea.[65] The most common grade 3 or 4 laboratory abnormalities include decreased neutrophil count and increased creatine phosphokinase.[65]
Mirdametinib was approved for medical use in the United States in February 2025.[63][65]
Selumetinib
Selumetinib is a drug produced by Astra Zeneca sold under the brand name Koselugo, and was approved by the FDA in April 2020[66] for the treatment of NF-1 in the pediatric population who are two or more years of age. It is a mitogen-activated protein kinase inhibitor (MEKi) and is indicated for use in pediatric patients who are symptomatic and have inoperable plexiform neurofibromas.[67] However, this medication is not curative and is not suitable for all patients.
Side effects of selumetinib include headache, nausea/vomiting, abdominal pain and other problems of the gastrointestinal tract, fatigue, muscle pain, constipation, paronychia as well as dry skin and other skin and hair problems.[68] The side effects can have a significant impact on a patient's life and may lead to someone having to discontinue treatment.
In an open-label, phase 2 trial of selumetinib with 50 children:
35 patients (70%) had a confirmed partial response, 28 of these patients had a durable response (lasting ≥1 year)
After 1 year of treatment, the mean decrease in child-reported tumour pain-intensity scores was 2 points, which is considered a clinically meaningful improvement
Prognosis
NF-1 is a progressive and diverse condition, making the prognosis difficult to predict. The NF-1 gene mutations manifest the disorder differently even amongst people of the same family. This phenomenon is called variable expressivity. For example, some individuals have almost no symptoms, while others may have a manifestation that is rapidly more progressive and can lead to significant disability and death.
For many NF-1 patients, a primary concern is the disfigurement caused by cutaneous/dermal neurofibromas, pigmented lesions, and the occasional limb abnormalities. However, there are many more severe complications caused by NF-1 like increased cancer risk, a plexiform neurofibroma has a 10-15% chance of developing into a MPNST (Malignant Peripheral Nerve Sheath Tumour) Epidemiology NF-1 is estimated to affect around 25,000 people in the UK.[69]
For many years, it was thought that Joseph Merrick (popularly known as the Elephant Man) had neurofibromatosis. In 1986, geneticists Tibbles and Cohen theorized that Merrick instead had the much rarer Proteus syndrome.[71]
↑Piersall, Linda, David H. Gutmann, & Rosalie Ferner. Living with Neurofibromatosis Type 1: A Guide for Adults. New York, NY: The National Neurofibromatosis Foundation, Inc. Print.
↑Hyman SL, Gill DS, Shores EA, Steinberg A, Joy P, Gibikote SV, etal. (April 2003). "Natural history of cognitive deficits and their relationship to MRI T2-hyperintensities in NF1". Neurology. 60 (7): 1139–1145. doi:10.1212/01.WNL.0000055090.78351.C1. PMID12682321. S2CID26812237.
↑Thompson HL, Viskochil DH, Stevenson DA, Chapman KL (February 2010). "Speech-language characteristics of children with neurofibromatosis type 1". American Journal of Medical Genetics. Part A. 152A (2): 284–290. doi:10.1002/ajmg.a.33235. PMID20101681. S2CID26650152.
↑Berardelli I, Maraone A, Belvisi D, Pasquini M, Giustini S, Miraglia E, etal. (November 2021). "The importance of suicide risk assessment in patients affected by neurofibromatosis". International Journal of Psychiatry in Clinical Practice. 25 (4): 350–355. doi:10.1080/13651501.2021.1921217. PMID34270353. S2CID235999769.
↑Brar KS, Trivedi C, Kaur N, Adnan M, Patel H, Beg U, etal. (October 2023). "Prevalence of Psychiatric Comorbidities in Patients With Neurofibromatosis". The Primary Care Companion for CNS Disorders. 25 (5): 49254. doi:10.4088/PCC.23m03514. PMID37816254. S2CID263240202.
12345678Garg S, Plasschaert E, Descheemaeker MJ, Huson S, Borghgraef M, Vogels A, etal. (2015-06-01). "Autism Spectrum Disorder Profile in Neurofibromatosis Type I". Journal of Autism and Developmental Disorders. 45 (6): 1649–1657. doi:10.1007/s10803-014-2321-5. ISSN1573-3432. PMID25475362.
↑Coltin H, Perreault S, Larouche V, Black K, Wilson B, Vanan MI, etal. (August 2022). "Selumetinib for symptomatic, inoperable plexiform neurofibromas in children with neurofibromatosis type 1: A national real-world case series". Pediatric Blood & Cancer. 69 (8) e29633. doi:10.1002/pbc.29633. PMID35289492. S2CID247453317.
↑Valeyrie-Allanore L, Roujeau JC, Revuz J (2005). "Neurofibromatosis type 1: From diagnosis to therapeutic challenges". American Journal of Medical Genetics Part A. 137A (1): 12–23. doi:10.1002/ajmg.a.38486. PMID29388340.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.





{kind=link}