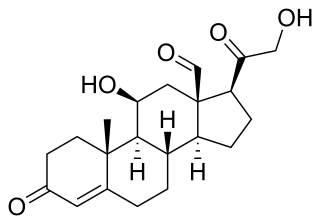
Primary aldosteronism (PA), also known as primary hyperaldosteronism, refers to the excess production of the hormone aldosterone from the adrenal glands, resulting in low renin levels and high blood pressure. This abnormality is a paraneoplastic syndrome. About 35% of the cases are caused by a single aldosterone-secreting adenoma, a condition known as Conn's syndrome.
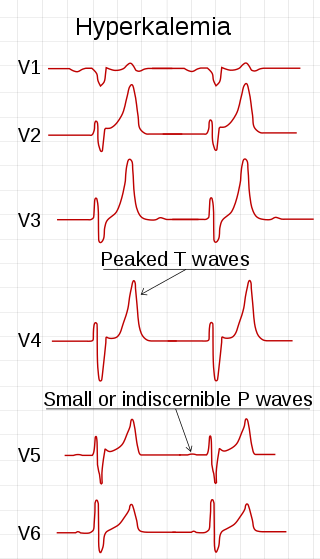
Hyperkalemia is an elevated level of potassium (K+) in the blood. Normal potassium levels are between 3.5 and 5.0 mmol/L (3.5 and 5.0 mEq/L) with levels above 5.5 mmol/L defined as hyperkalemia. Typically hyperkalemia does not cause symptoms. Occasionally when severe it can cause palpitations, muscle pain, muscle weakness, or numbness. Hyperkalemia can cause an abnormal heart rhythm which can result in cardiac arrest and death.

Amiloride, sold under the trade name Midamor among others, is a medication typically used with other medications to treat high blood pressure or swelling due to heart failure or cirrhosis of the liver. Amiloride is classified as a potassium-sparing diuretic. Amiloride is often used together with another diuretic, such as a thiazide or loop diuretic. It is taken by mouth. Onset of action is about two hours and it lasts for about a day.
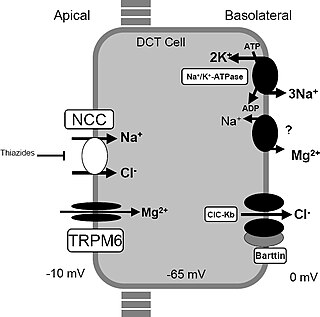
Gitelman syndrome (GS) is an autosomal recessive kidney tubule disorder characterized by low blood levels of potassium and magnesium, decreased excretion of calcium in the urine, and elevated blood pH. It is the most frequent hereditary salt-losing tubulopathy. Gitelman syndrome is caused by disease-causing variants on both alleles of the SLC12A3 gene. The SLC12A3 gene encodes the thiazide-sensitive sodium-chloride cotransporter, which can be found in the distal convoluted tubule of the kidney.

Potassium-sparing diuretics or antikaliuretics refer to drugs that cause diuresis without causing potassium loss in the urine. They are typically used as an adjunct in management of hypertension, cirrhosis, and congestive heart failure. The steroidal aldosterone antagonists can also be used for treatment of primary hyperaldosteronism. Spironolactone, a steroidal aldosterone antagonist, is also used in management of female hirsutism and acne from PCOS or other causes.
Hypoaldosteronism is an endocrinological disorder characterized by decreased levels of the hormone aldosterone. Similarly, isolated hypoaldosteronism is the condition of having lowered aldosterone without corresponding changes in cortisol.
Secondary hypertension is a type of hypertension which has a specific and identifiable underlying primary cause. It is much less common than essential hypertension, affecting only 5-10% of hypertensive patients. It has many different causes including obstructive sleep apnea, kidney disease, endocrine diseases, and tumors. The cause of secondary hypertension varies significantly with age. It also can be a side effect of many medications.

Metabolic alkalosis is an acid-base disorder in which the pH of tissue is elevated beyond the normal range (7.35–7.45). This is the result of decreased hydrogen ion concentration, leading to increased bicarbonate, or alternatively a direct result of increased bicarbonate concentrations. The condition typically cannot last long if the kidneys are functioning properly.

Renal tubular acidosis (RTA) is a medical condition that involves an accumulation of acid in the body due to a failure of the kidneys to appropriately acidify the urine. In renal physiology, when blood is filtered by the kidney, the filtrate passes through the tubules of the nephron, allowing for exchange of salts, acid equivalents, and other solutes before it drains into the bladder as urine. The metabolic acidosis that results from RTA may be caused either by insufficient secretion of hydrogen ions into the latter portions of the nephron or by failure to reabsorb sufficient bicarbonate ions from the filtrate in the early portion of the nephron. Although a metabolic acidosis also occurs in those with chronic kidney disease, the term RTA is reserved for individuals with poor urinary acidification in otherwise well-functioning kidneys. Several different types of RTA exist, which all have different syndromes and different causes. RTA is usually an incidental finding based on routine blood draws that show abnormal results. Clinically, patients may present with vague symptoms such as dehydration, mental status changes, or delayed growth in adolescents.

Apparent mineralocorticoid excess is an autosomal recessive disorder causing hypertension, hypernatremia and hypokalemia. It results from mutations in the HSD11B2 gene, which encodes the kidney isozyme of 11β-hydroxysteroid dehydrogenase type 2. In an unaffected individual, this isozyme inactivates circulating cortisol to the less active metabolite cortisone. The inactivating mutation leads to elevated local concentrations of cortisol in the aldosterone sensitive tissues like the kidney. Cortisol at high concentrations can cross-react and activate the mineralocorticoid receptor due to the non-selectivity of the receptor, leading to aldosterone-like effects in the kidney. This is what causes the hypokalemia, hypertension, and hypernatremia associated with the syndrome. Patients often present with severe hypertension and end-organ changes associated with it like left ventricular hypertrophy, retinal, renal and neurological vascular changes along with growth retardation and failure to thrive. In serum both aldosterone and renin levels are low.

Liddle's syndrome, also called Liddle syndrome, is a genetic disorder inherited in an autosomal dominant manner that is characterized by early, and frequently severe, high blood pressure associated with low plasma renin activity, metabolic alkalosis, low blood potassium, and normal to low levels of aldosterone. Liddle syndrome involves abnormal kidney function, with excess reabsorption of sodium and loss of potassium from the renal tubule, and is treated with a combination of low sodium diet and potassium-sparing diuretics. It is extremely rare, with fewer than 30 pedigrees or isolated cases having been reported worldwide as of 2008.

Oculocerebrorenal syndrome is a rare X-linked recessive disorder characterized by congenital cataracts, hypotonia, intellectual disability, proximal tubular acidosis, aminoaciduria and low-molecular-weight proteinuria. Lowe syndrome can be considered a cause of Fanconi syndrome.
Pseudohyperaldosteronism is a medical condition which mimics the effects of elevated aldosterone (hyperaldosteronism) by presenting with high blood pressure, low blood potassium levels (hypokalemia), metabolic alkalosis, and low levels of plasma renin activity (PRA). However, unlike hyperaldosteronism, this conditions exhibits low or normal levels of aldosterone in the blood. Causes include genetic disorders, acquired conditions, metabolic disorders, and dietary imbalances including excessive consumption of licorice. Confirmatory diagnosis depends on the specific cause and may involve blood tests, urine tests, or genetic testing; however, all forms of this condition exhibit abnormally low concentrations of both plasma renin activity (PRA) and plasma aldosterone concentration (PAC) which differentiates this group of conditions from other forms of secondary hypertension. Treatment is tailored to the specific cause and focuses on symptom control, blood pressure management, and avoidance of triggers.

The epithelial sodium channel(ENaC), (also known as amiloride-sensitive sodium channel) is a membrane-bound ion channel that is selectively permeable to sodium ions (Na+). It is assembled as a heterotrimer composed of three homologous subunits α or δ, β, and γ, These subunits are encoded by four genes: SCNN1A, SCNN1B, SCNN1G, and SCNN1D. The ENaC is involved primarily in the reabsorption of sodium ions at the collecting ducts of the kidney's nephrons. In addition to being implicated in diseases where fluid balance across epithelial membranes is perturbed, including pulmonary edema, cystic fibrosis, COPD and COVID-19, proteolyzed forms of ENaC function as the human salt taste receptor.

The sodium-chloride symporter (also known as Na+-Cl− cotransporter, NCC or NCCT, or as the thiazide-sensitive Na+-Cl− cotransporter or TSC) is a cotransporter in the kidney which has the function of reabsorbing sodium and chloride ions from the tubular fluid into the cells of the distal convoluted tubule of the nephron. It is a member of the SLC12 cotransporter family of electroneutral cation-coupled chloride cotransporters. In humans, it is encoded by the SLC12A3 gene (solute carrier family 12 member 3) located in 16q13.

The SCNN1B gene encodes for the β subunit of the epithelial sodium channel ENaC in vertebrates. ENaC is assembled as a heterotrimer composed of three homologous subunits α, β, and γ or δ, β, and γ. The other ENAC subunits are encoded by SCNN1A, SCNN1G, and SCNN1D.

The SCNN1A gene encodes for the α subunit of the epithelial sodium channel ENaC in vertebrates. ENaC is assembled as a heterotrimer composed of three homologous subunits α, β, and γ or δ, β, and γ. The other ENAC subunits are encoded by SCNN1B, SCNN1G, and SCNN1D.
The SCNN1G gene encodes for the γ subunit of the epithelial sodium channel ENaC in vertebrates. ENaC is assembled as a heterotrimer composed of three homologous subunits α, β, and γ or δ, β, and γ. The other ENAC subunits are encoded by SCNN1A, SCNN1B, and SCNN1D.

Serine/threonine protein kinase WNK4 also known as With No lysine (K) protein kinase 4(WNK4), is an enzyme that in humans is encoded by the WNK4 gene. Missense mutations cause a genetic form of pseudohypoaldosteronism type 2, also called Gordon syndrome or Familial Hyperkalemic Hypertension.

Distal renal tubular acidosis (dRTA) is the classical form of RTA, being the first described. Distal RTA is characterized by a failure of acid secretion by the alpha intercalated cells of the distal tubule and cortical collecting duct of the distal nephron. This failure of acid secretion may be due to a number of causes. It leads to relatively alkaline urine, due to the kidney's inability to acidify the urine to a pH of less than 5.3.
