
A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Neurofibromatosis (NF) is a group of three conditions in which tumors grow in the nervous system. The three types are neurofibromatosis type I (NF1), neurofibromatosis type II (NF2), and schwannomatosis. In NF1 symptoms include light brown spots on the skin, freckles in the armpit and groin, small bumps within nerves, and scoliosis. In NF2, there may be hearing loss, cataracts at a young age, balance problems, flesh colored skin flaps, and muscle wasting. In schwannomatosis there may be pain either in one location or in wide areas of the body. The tumors in NF are generally non-cancerous.

Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

Noonan syndrome (NS) is a genetic disorder that may present with mildly unusual facial features, short height, congenital heart disease, bleeding problems, and skeletal malformations. Facial features include widely spaced eyes, light-colored eyes, low-set ears, a short neck, and a small lower jaw. Heart problems may include pulmonary valve stenosis. The breast bone may either protrude or be sunken, while the spine may be abnormally curved. Intelligence in the syndrome is often normal. Complications of NS can include leukemia.

Lisch nodule, also known as iris hamartoma, is a pigmented hamartomatous nodular aggregate of dendritic melanocytes affecting the iris, named after Austrian ophthalmologist Karl Lisch (1907–1999), who first recognized them in 1937.

Café au lait spots, or café au lait macules, are flat, hyperpigmented birthmarks. The name café au lait is French for "coffee with milk" and refers to their light-brown color. Café au lait lesions with rough borders may be seen in McCune-Albright syndrome. In contrast, Café au lait lesions of neurofibromatosis have smooth borders.

Neurofibromatosis type I (NF-1) is a complex multi-system human disorder caused by the mutation of a gene on chromosome 17 that is responsible for production of a protein, called neurofibromin, which is needed for normal function in many human cell types. NF-1 causes tumors along the nervous system which can grow anywhere on the body. NF-1 is one of the most common genetic disorders and is not limited to any person's race or sex. NF-1 is an autosomal dominant disorder, which means that mutation or deletion of one copy of the NF-1 gene is sufficient for the development of NF-1, although presentation varies widely and is often different even between relatives affected by NF-1.
Neurofibromatosis type II is a genetic condition which may be inherited or may arise spontaneously. The main manifestation of the condition is the development of bilateral benign brain tumors in the nerve sheath of the cranial nerve VIII, which is the "auditory-vestibular nerve" that transmits sensory information from the inner ear to the brain. Besides, other benign brain and spinal tumors occur. Symptoms depend on the presence, localisation and growth of the tumor(s). Many people with this condition also experience visual problems. Neurofibromatosis type II is caused by mutations of the "Merlin" gene, which seems to influence the form and movement of cells. The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions. Historically the underlying disorder has not had any therapy due to the cell function caused by the genetic mutation.

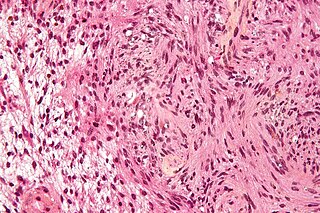
A neurofibroma is a benign nerve-sheath tumor in the peripheral nervous system. In 90% of cases, they are found as stand-alone tumors, while the remainder are found in persons with neurofibromatosis type I (NF1), an autosomal-dominant genetically inherited disease. They can result in a range of symptoms from physical disfiguration and pain to cognitive disability.
Phakomatoses, or phacomatosis pigmentovascularis (PPV), is the term used for a group of rare syndromes involving structures arising from the embryonic ectoderm. These are characterised by vascular and pigmentary birthmarks or skin lesions, and often involving multiple organ systems in the body. The term is used to describe the “association of a vascular nevus with an extensive pigmentary nevus”.

Hypertelorism is an abnormally increased distance between two organs or bodily parts, usually referring to an increased distance between the orbits (eyes), or orbital hypertelorism. In this condition the distance between the inner eye corners as well as the distance between the pupils is greater than normal. Hypertelorism should not be confused with telecanthus, in which the distance between the inner eye corners is increased but the distances between the outer eye corners and the pupils remain unchanged.
The Crowe sign or Crowe's sign is the presence of axillary (armpit) freckling in people with neurofibromatosis type I. These freckles occur in up to 30% of people with the disease and their presence is one of six diagnostic criteria for neurofibromatosis. Freckles can also be present in the intertriginous area in neurofibromatosis, such as the inguinal fold, submamillary areas and nape of the neck.
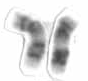
Chromosome 17 is one of the 23 pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 17 spans more than 83 million base pairs and represents between 2.5 and 3% of the total DNA in cells.

Noonan syndrome with multiple lentigines (NSML) which is part of a group called Ras/MAPK pathway syndromes, is a rare autosomal dominant, multisystem disease caused by a mutation in the protein tyrosine phosphatase, non-receptor type 11 gene (PTPN11). The disease is a complex of features, mostly involving the skin, skeletal and cardiovascular systems, which may or may not be present in all patients. The nature of how the mutation causes each of the condition's symptoms is not well known; however, research is ongoing. It is a RASopathy.

Neurofibromin 1 (NF1) is a gene in humans that is located on chromosome 17. NF1 codes for neurofibromin, a GTPase-activating protein that negatively regulates RAS/MAPK pathway activity by accelerating the hydrolysis of Ras-bound GTP. NF1 has a high mutation rate and mutations in NF1 can alter cellular growth control, and neural development, resulting in neurofibromatosis type 1. Symptoms of NF1 include disfiguring cutaneous neurofibromas (CNF), café au lait pigment spots, plexiform neurofibromas (PN), skeletal defects, optic nerve gliomas, life-threatening malignant peripheral nerve sheath tumors (MPNST), pheochromocytoma, attention deficits, learning deficits and other cognitive disabilities.
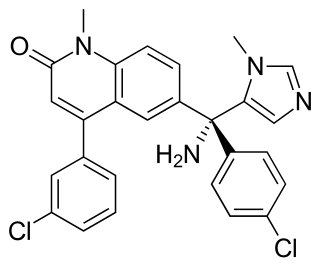
Tipifarnib is a farnesyltransferase inhibitor. Farnesyltransferase inhibitors block the activity of the farnesyltransferase enzyme by inhibiting prenylation of the CAAX tail motif, which ultimately prevents Ras from binding to the membrane, rendering it inactive.

Legius syndrome (LS) is an autosomal dominant condition characterized by cafe au lait spots. It was first described in 2007 and is often mistaken for neurofibromatosis type I (NF-1). It is caused by mutations in the SPRED1 gene. It is also known as neurofibromatosis type 1-like syndrome (NFLS).
The RASopathies are developmental syndromes caused by germline mutations in genes that alter the Ras subfamily and mitogen-activated protein kinases that control signal transduction, including:
Microdeletion syndrome is a syndrome caused by a chromosomal deletion smaller than 5 million base pairs spanning several genes that is too small to be detected by conventional cytogenetic methods or high resolution karyotyping. Detection is done by fluorescence in situ hybridization (FISH). Larger chromosomal deletion syndromes are detectable using karyotyping techniques.