A congenital disorder of glycosylation is one of several rare inborn errors of metabolism in which glycosylation of a variety of tissue proteins and/or lipids is deficient or defective. Congenital disorders of glycosylation are sometimes known as CDG syndromes. They often cause serious, sometimes fatal, malfunction of several different organ systems in affected infants. The most common sub-type is PMM2-CDG where the genetic defect leads to the loss of phosphomannomutase 2 (PMM2), the enzyme responsible for the conversion of mannose-6-phosphate into mannose-1-phosphate.
Lipodystrophy syndromes are a group of genetic or acquired disorders in which the body is unable to produce and maintain healthy fat tissue. The medical condition is characterized by abnormal or degenerative conditions of the body's adipose tissue. A more specific term, lipoatrophy, is used when describing the loss of fat from one area. This condition is also characterized by a lack of circulating leptin which may lead to osteosclerosis. The absence of fat tissue is associated with insulin resistance, hypertriglyceridemia, non-alcoholic fatty liver disease (NAFLD) and metabolic syndrome.

Chylomicrons, also known as ultra low-density lipoproteins (ULDL), are lipoprotein particles that consist of triglycerides (85–92%), phospholipids (6–12%), cholesterol (1–3%), and proteins (1–2%). They transport dietary lipids, such as fats and cholesterol, from the intestines to other locations in the body, within the water-based solution of the bloodstream. ULDLs are one of the five major groups lipoproteins are divided into based on their density. A protein specific to chylomicrons is ApoB48.

Lipomatosis is believed to be an autosomal dominant condition in which multiple lipomas are present on the body. Many discrete, encapsulated lipomas form on the trunk and extremities, with relatively few on the head and shoulders. In 1993, a genetic polymorphism within lipomas was localized to chromosome 12q15, where the HMGIC gene encodes the high-mobility-group protein isoform I-C. This is one of the most commonly found mutations in solitary lipomatous tumors but lipomas often have multiple mutations. Reciprocal translocations involving chromosomes 12q13 and 12q14 have also been observed within.

Laminopathies are a group of rare genetic disorders caused by mutations in genes encoding proteins of the nuclear lamina. Since the first reports of laminopathies in the late 1990s, increased research efforts have started to uncover the vital role of nuclear envelope proteins in cell and tissue integrity in animals. Laminopathies are a group of degenerative diseases, other disorders associated with inner nuclear membrane proteins are known as nuclear envelopathies.
Barraquer–Simons syndrome is a rare form of lipodystrophy, which usually first affects the head, and then spreads to the thorax. It is named for Luis Barraquer Roviralta (1855–1928), a Spanish physician, and Arthur Simons (1879–1942), a German physician. Some evidence links it to LMNB2.

Seipin is a protein that in humans is encoded by the BSCL2 gene.

1-acyl-sn-glycerol-3-phosphate acyltransferase beta is an enzyme that in humans is encoded by the AGPAT2 gene.

Neutral lipid storage disease is a congenital autosomal recessive disorder characterized by accumulation of triglycerides in the cytoplasm of leukocytes, muscle, liver, fibroblasts, and other tissues. It commonly occurs as one of two subtypes, cardiomyopathic neutral lipid storage disease (NLSD-M), or ichthyotic neutral lipid storage disease (NLSD-I) which is also known as Chanarin–Dorfman syndrome), which are characterized primarily by myopathy and ichthyosis, respectively. Normally, the ichthyosis that is present is typically non-bullous congenital ichthyosiform erythroderma which appears as white scaling.
Acquired generalized lipodystrophy (AGL), also known as Lawrence syndrome and Lawrence–Seip syndrome, is a rare skin condition that appears during childhood or adolescence, characterized by fat loss affecting large areas of the body, particularly the face, arms, and legs. There are four types of lipodystrophy based on its onset and areas affected: acquired or inherited, and generalized or partial. Both acquired or inherited lipodystrophy present as loss of adipose tissues, in the absence of nutritional deprivation. The near-total loss of subcutaneous adipose tissue is termed generalized lipodystrophy while the selective loss of adipose tissues is denoted as partial lipodystrophy. Thus, as the name suggests, AGL is a near-total deficiency of adipose tissues in the body that is developed later in life. It is an extremely rare disease with only about 100 cases reported worldwide. There are three main etiologies of AGL suspected: autoimmune, panniculitis-associated, or idiopathic. After its onset, the disease progresses over a few days, weeks, months, or even in years. Clinical presentations of AGL are similar to other lipodystrophies, including metabolic complications and hypoleptinemia. Treatments are also similar and mainly supportive for symptomatic alleviation. Although HIV- or drug-induced lipodystrophy are types of acquired lipodystrophy, their origins are very specific and distinct and hence are usually not discussed with AGL.
Familial partial lipodystrophy, also known as Köbberling–Dunnigan syndrome, is a rare genetic metabolic condition characterized by the loss of subcutaneous fat.
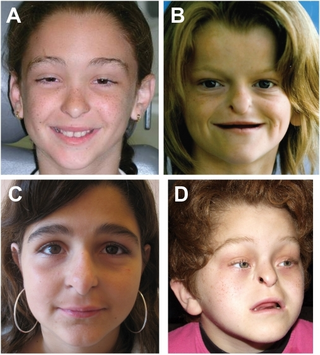
Johanson–Blizzard syndrome (JBS) is a rare, sometimes fatal autosomal recessive multisystem congenital disorder featuring abnormal development of the pancreas, nose and scalp, with intellectual disability, hearing loss and growth failure. It is sometimes described as a form of ectodermal dysplasia.

Cantú syndrome is a rare condition characterized by hypertrichosis, osteochondrodysplasia, and cardiomegaly. Less than 50 cases have been described in the literature; they are associated with a mutation in the ABCC9-gene that codes for the ABCC9-protein.
Progeroid syndromes (PS) are a group of rare genetic disorders that mimic physiological aging, making affected individuals appear to be older than they are. The term progeroid syndrome does not necessarily imply progeria, which is a specific type of progeroid syndrome.

Wiedemann–Rautenstrauch (WR) syndrome, also known as neonatal progeroid syndrome, is a rare autosomal recessive progeroid syndrome. There have been over 30 cases of WR. WR is associated with abnormalities in bone maturation, and lipids and hormone metabolism.
Metreleptin, sold under the brand name Myalept among others, is a synthetic analog of the hormone leptin used to treat various forms of dyslipidemia. It has been approved in Japan for metabolic disorders including lipodystrophy and in the United States as replacement therapy to treat the complications of leptin deficiency, in addition to diet, in patients with congenital generalized or acquired generalized lipodystrophy.
Seipin is a homo-oligomeric integral membrane protein in the endoplasmic reticulum (ER) that concentrates at junctions with cytoplasmic lipid droplets (LDs). Alternatively, seipin can be referred to as Berardinelli–Seip congenital lipodystrophy type 2 protein (BSCL2), and it is encoded by the corresponding gene of the same name, i.e. BSCL2. At protein level, seipin is expressed in cortical neurons in the frontal lobes, as well as motor neurons in the spinal cord. It is highly expressed in areas like the brain, testis and adipose tissue. Seipin's function is still unclear but it has been localized close to lipid droplets, and cells knocked out in seipin have anomalous droplets. Hence, recent evidence suggests that seipin plays a crucial role in lipid droplet biogenesis.
Asprosin is a protein hormone produced by mammals in tissues that stimulates the liver to release glucose into the blood stream. Asprosin is encoded by the gene FBN1 as part of the protein profibrillin and is released from the C-terminus of the latter by specific proteolysis. In the liver, asprosin activates rapid glucose release via a cyclic adenosine monophosphate (cAMP)-dependent pathway.

Marfanoid–progeroid–lipodystrophy syndrome (MPL), also known as Marfan lipodystrophy syndrome (MFLS) or progeroid fibrillinopathy, is an extremely rare medical condition which manifests as a variety of symptoms including those usually associated with Marfan syndrome, an appearance resembling that seen in neonatal progeroid syndrome, and severe partial lipodystrophy. It is a genetic condition that is caused by mutations in the FBN1 gene, which encodes profibrillin, and affects the cleavage products of profibrillin, fibrillin-1, a fibrous structural protein, and asprosin, a glucogenic protein hormone. As of 2016, fewer than 10 cases of the condition have been reported. Lizzie Velásquez and Abby Solomon have become known publicly through the media for having the condition.

Facial infiltrating lipomatosis (FIL), also referred to as congenital infiltrating lipomatosis of the face or facial infused lipomatosis, is an ultra-rare craniofacial overgrowth condition caused by a genetic mutation of the PIK3CA gene. The condition is a part of the PIK3CA related overgrowth spectrum (PROS). The disease is congenital and non-hereditary. First described by Slavin and colleagues in 1983.
