Related Research Articles

Metabolic syndrome is a clustering of at least three of the following five medical conditions: abdominal obesity, high blood pressure, high blood sugar, high serum triglycerides, and low serum high-density lipoprotein (HDL).

Leptin is a protein hormone predominantly made by adipocytes and its primary role is likely to regulate long-term energy balance.
Lipodystrophy syndromes are a group of genetic or acquired disorders in which the body is unable to produce and maintain healthy fat tissue. The medical condition is characterized by abnormal or degenerative conditions of the body's adipose tissue. A more specific term, lipoatrophy, is used when describing the loss of fat from one area. This condition is also characterized by a lack of circulating leptin which may lead to osteosclerosis. The absence of fat tissue is associated with insulin resistance, hypertriglyceridemia, non-alcoholic fatty liver disease (NAFLD) and metabolic syndrome.

Lipolysis is the metabolic pathway through which lipid triglycerides are hydrolyzed into a glycerol and free fatty acids. It is used to mobilize stored energy during fasting or exercise, and usually occurs in fat adipocytes. The most important regulatory hormone in lipolysis is insulin; lipolysis can only occur when insulin action falls to low levels, as occurs during fasting. Other hormones that affect lipolysis include leptin, glucagon, epinephrine, norepinephrine, growth hormone, atrial natriuretic peptide, brain natriuretic peptide, and cortisol.

Adipose tissue is a loose connective tissue composed mostly of adipocytes. In addition to adipocytes, adipose tissue contains the stromal vascular fraction (SVF) of cells including preadipocytes, fibroblasts, vascular endothelial cells and a variety of immune cells such as adipose tissue macrophages. Adipose tissue is derived from preadipocytes. Its main role is to store energy in the form of lipids, although it also cushions and insulates the body. Far from being hormonally inert, adipose tissue has, in recent years, been recognized as a major endocrine organ, as it produces hormones such as leptin, estrogen, resistin, and cytokines. In obesity, adipose tissue is also implicated in the chronic release of pro-inflammatory markers known as adipokines, which are responsible for the development of metabolic syndrome, a constellation of diseases, including type 2 diabetes, cardiovascular disease and atherosclerosis. The two types of adipose tissue are white adipose tissue (WAT), which stores energy, and brown adipose tissue (BAT), which generates body heat. The formation of adipose tissue appears to be controlled in part by the adipose gene. Adipose tissue – more specifically brown adipose tissue – was first identified by the Swiss naturalist Conrad Gessner in 1551.

In pharmacology, the fibrates are a class of amphipathic carboxylic acids and esters. They are derivatives of fibric acid. They are used for a range of metabolic disorders, mainly hypercholesterolemia, and are therefore hypolipidemic agents.

Adipocytes, also known as lipocytes and fat cells, are the cells that primarily compose adipose tissue, specialized in storing energy as fat. Adipocytes are derived from mesenchymal stem cells which give rise to adipocytes through adipogenesis. In cell culture, adipocyte progenitors can also form osteoblasts, myocytes and other cell types.

Hypertriglyceridemia is the presence of high amounts of triglycerides in the blood. Triglycerides are the most abundant fatty molecule in most organisms. Hypertriglyceridemia occurs in various physiologic conditions and in various diseases, and high triglyceride levels are associated with atherosclerosis, even in the absence of hypercholesterolemia and predispose to cardiovascular disease.

Panniculitis is a group of diseases whose hallmark is inflammation of subcutaneous adipose tissue. Symptoms include tender skin nodules, and systemic signs such as weight loss and fatigue.
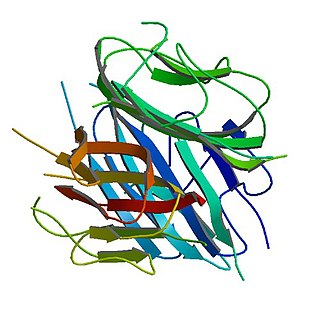
Adiponectin is a protein hormone and adipokine, which is involved in regulating glucose levels and fatty acid breakdown. In humans, it is encoded by the ADIPOQ gene and is produced primarily in adipose tissue, but also in muscle and even in the brain.
In biochemistry, lipogenesis is the conversion of fatty acids and glycerol into fats, or a metabolic process through which acetyl-CoA is converted to triglyceride for storage in fat. Lipogenesis encompasses both fatty acid and triglyceride synthesis, with the latter being the process by which fatty acids are esterified to glycerol before being packaged into very-low-density lipoprotein (VLDL). Fatty acids are produced in the cytoplasm of cells by repeatedly adding two-carbon units to acetyl-CoA. Triacylglycerol synthesis, on the other hand, occurs in the endoplasmic reticulum membrane of cells by bonding three fatty acid molecules to a glycerol molecule. Both processes take place mainly in liver and adipose tissue. Nevertheless, it also occurs to some extent in other tissues such as the gut and kidney. A review on lipogenesis in the brain was published in 2008 by Lopez and Vidal-Puig. After being packaged into VLDL in the liver, the resulting lipoprotein is then secreted directly into the blood for delivery to peripheral tissues.
Lipoatrophy is the term describing the localized loss of fat tissue. This may occur as a result of subcutaneous injections of insulin in the treatment of diabetes, from the use of human growth hormone or from subcutaneous injections of copaxone used for the treatment of multiple sclerosis. In the latter case, an injection may produce a small dent at the injection site. Lipoatrophy occurs in HIV-associated lipodystrophy, one cause of which is an adverse drug reaction that is associated with some antiretroviral medications.
Barraquer–Simons syndrome is a rare form of lipodystrophy, which usually first affects the head, and then spreads to the thorax. It is named for Luis Barraquer Roviralta (1855–1928), a Spanish physician, and Arthur Simons (1879–1942), a German physician. Some evidence links it to LMNB2.
Congenital generalized lipodystrophy is an extremely rare autosomal recessive condition, characterized by an extreme scarcity of fat in the subcutaneous tissues. It is a type of lipodystrophy disorder where the magnitude of fat loss determines the severity of metabolic complications. Only 250 cases of the condition have been reported, and it is estimated that it occurs in 1 in 10 million people worldwide.
Familial partial lipodystrophy, also known as Köbberling–Dunnigan syndrome, is a rare genetic metabolic condition characterized by the loss of subcutaneous fat.

Familial hypertriglyceridemia is a genetic disorder characterized by the liver overproducing very-low-density lipoproteins (VLDL). As a result, an affected individual will have an excessive number of VLDL and triglycerides on a lipid profile. This genetic disorder usually follows an autosomal dominant inheritance pattern. The disorder presents clinically in patients with mild to moderate elevations in triglyceride levels. Familial hypertriglyceridemia is typically associated with other co-morbid conditions such as hypertension, obesity, and hyperglycemia. Individuals with the disorder are mostly heterozygous in an inactivating mutation of the gene encoding for lipoprotein lipase (LPL). This sole mutation can markedly elevate serum triglyceride levels. However, when combined with other medications or pathologies it can further elevate serum triglyceride levels to pathologic levels. Substantial increases in serum triglyceride levels can lead to certain clinical signs and the development of acute pancreatitis.

Lipotoxicity is a metabolic syndrome that results from the accumulation of lipid intermediates in non-adipose tissue, leading to cellular dysfunction and death. The tissues normally affected include the kidneys, liver, heart and skeletal muscle. Lipotoxicity is believed to have a role in heart failure, obesity, and diabetes, and is estimated to affect approximately 25% of the adult American population.
MDP syndrome, also known as mandibular dysplasia with deafness and progeroid features, is an extremely rare metabolic disorder that prevents fatty tissue from being stored underneath the skin. It is only known to affect a very small number of people worldwide. Recent research has suggested that it may be caused by an abnormality of the POLD1 gene on chromosome 19, which causes an enzyme crucial to DNA replication to be defective.
Metreleptin, sold under the brand name Myalept among others, is a synthetic analog of the hormone leptin used to treat various forms of dyslipidemia. It has been approved in Japan for metabolic disorders including lipodystrophy and in the United States as replacement therapy to treat the complications of leptin deficiency, in addition to diet, in patients with congenital generalized or acquired generalized lipodystrophy.
The Metabolic Score for Insulin Resistance (METS-IR) is a metabolic index developed with the aim to quantify peripheral insulin sensitivity in humans; it was first described under the name METS-IR by Bello-Chavolla et al. in 2018. It was developed by the Metabolic Research Disease Unit at the Instituto Nacional de Ciencias Médicas Salvador Zubirán and validated against the euglycemic hyperinsulinemic clamp and the frequently-sampled intravenous glucose tolerance test in Mexican population. It is a non-insulin-based alternative to insulin-based methods to quantify peripheral insulin sensitivity and an alternative to the Homeostatic Model Assessment (HOMA-IR) and the quantitative insulin sensitivity check index (QUICKI). METS-IR is currently validated for its use to assess cardio-metabolic risk in Latino population.
References
- 1 2 Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L., eds. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.[ page needed ]
- ↑ James, William D.; Berger, Timothy G.; Elston, Dirk M., eds. (2006). Andrews' Diseases of the Skin: Clinical Dermatology (Ninth ed.). Saunders Elsevier. ISBN 978-0-7216-2921-6.
- 1 2 Brown, Rebecca J.; Chan, Jean L.; Jaffe, Elaine S.; Cochran, Elaine; DePaoli, Alex M.; Gautier, Jean-Francois; Goujard, Cecile; Vigouroux, Corinne; Gorden, Phillip (2016-01-02). "Lymphoma in acquired generalized lipodystrophy". Leukemia & Lymphoma. 57 (1): 45–50. doi:10.3109/10428194.2015.1040015. PMC 4755279 . PMID 25864863.
- ↑ Garg, Abhimanyu (2004-03-18). "Acquired and Inherited Lipodystrophies". New England Journal of Medicine. 350 (12): 1220–1234. doi:10.1056/nejmra025261. PMID 15028826.
- 1 2 3 4 5 6 7 8 Vantyghem, Pr Marie-Christine, ed. (January 2009). "Orphanet: Acquired generalized lipodystrophy". www.orpha.net. Retrieved 2017-11-07.
- 1 2 3 4 5 6 7 8 9 10 11 Hussain, Iram; Garg, Abhimanyu (2016). "Lipodystrophy Syndromes". Endocrinology and Metabolism Clinics of North America. 45 (4): 783–797. doi:10.1016/j.ecl.2016.06.012. PMC 4947059 . PMID 27823605.
- 1 2 3 4 5 6 7 8 9 Handelsman, Yehuda; Oral, Elif A.; Bloomgarden, Zachary T.; Brown, Rebecca J.; Chan, Jean L.; Einhorn, Daniel; Garber, Alan J.; Garg, Abhimanyu; Garvey, W. Timothy (2013). "THE CLINICAL APPROACH TO THE DETECTION OF LIPODYSTROPHY – AN AACE CONSENSUS STATEMENT". Endocrine Practice. 19 (1): 107–116. doi:10.4158/endp.19.1.v767575m65p5mr06. PMC 4108221 . PMID 23435042.
- 1 2 3 Gardner, David G.; Shoback, Dolores M. (2017-10-10). Gardner, David G.; Shoback, Dolores M. (eds.). Greenspan's Basic & Clinical Endocrinology (Tenth ed.). New York: McGraw-Hill Education Lange. ISBN 9781259589287. OCLC 995848612.
- 1 2 Musso, Carla; Major, Maria Laura; Andres, Eugenia; Simha, Vinaya (2017-01-05). "Metreleptin Treatment in Three Patients with Generalized Lipodystrophy". Clinical Medicine Insights: Case Reports. 9: 123–127. doi:10.4137/ccrep.s40196. PMC 5217977 . PMID 28096701.
- ↑ Billings, J. K.; Milgraum, S. S.; Gupta, A. K.; Headington, J. T.; Rasmussen, J. E. (December 1987). "Lipoatrophic panniculitis: a possible autoimmune inflammatory disease of fat. Report of three cases". Archives of Dermatology. 123 (12): 1662–1666. doi:10.1001/archderm.123.12.1662. PMID 3688906.
- 1 2 3 4 5 6 Nolis, Tom (2013). "Exploring the pathophysiology behind the more common genetic and acquired lipodystrophies". Journal of Human Genetics. 59 (1): 16–23. doi: 10.1038/jhg.2013.107 . PMID 24152769.
- 1 2 3 4 5 Oral, Elif Arioglu; Simha, Vinaya; Ruiz, Elaine; Andewelt, Alexa; Premkumar, Ahalya; Snell, Peter; Wagner, Anthony J.; DePaoli, Alex M.; Reitman, Marc L. (2002-02-21). "Leptin-Replacement Therapy for Lipodystrophy". New England Journal of Medicine. 346 (8): 570–578. doi: 10.1056/nejmoa012437 . PMID 11856796.
- ↑ Hübler, A.; Abendroth, K.; Keiner, T.; Stöcker, W.; Kauf, E.; Hein, G.; Stein, G. (1998). "Dysregulation of insulin-like growth factors in a case of generalized acquired lipoatrophic diabetes mellitus (Lawrence Syndrome) connected with autoantibodies against adipocyte membranes". Experimental and Clinical Endocrinology & Diabetes. 106 (1): 79–84. doi:10.1055/s-0029-1211955. PMID 9516065.
- 1 2 3 4 5 "Homepage | Myalept". www.myalept.com. Retrieved 2017-11-07.
- ↑ Bolan, Charles; Oral, Elif Arioglu; Gorden, Phillip; Taylor, Simeon; Leitman, Susan F. (January 2002). "Intensive, long-term plasma exchange therapy for severe hypertriglyceridemia in acquired generalized lipoatrophy". The Journal of Clinical Endocrinology and Metabolism. 87 (1): 380–384. doi: 10.1210/jcem.87.1.8176 . PMID 11788680.
- 1 2 Diker-Cohen, Talia; Cochran, Elaine; Gorden, Phillip; Brown, Rebecca J. (2015-05-01). "Partial and Generalized Lipodystrophy: Comparison of Baseline Characteristics and Response to Metreleptin". The Journal of Clinical Endocrinology & Metabolism. 100 (5): 1802–1810. doi:10.1210/jc.2014-4491. PMC 4422900 . PMID 25734254.
- ↑ Cunningham, Julia; Nadal, Rosa; Broome, Catherine (2017). "Acquired Generalized Lipodystrophy Following Immune Thrombocytopenia". The American Journal of Medicine. 130 (10): e445–e446. doi: 10.1016/j.amjmed.2017.04.035 . PMID 28549922.
- ↑ Patni, Nivedita; Xing, Chao; Agarwal, Anil K.; Garg, Abhimanyu (2017-09-01). "Juvenile-onset generalized lipodystrophy due to a novel heterozygous missense LMNA mutation affecting lamin C". American Journal of Medical Genetics Part A. 173 (9): 2517–2521. doi:10.1002/ajmg.a.38341. PMC 5593256 . PMID 28686329.