Type 2 diabetes, sleep apnea, high blood pressure,[3] high cholesterol, heart problems, particularly enlargement of the heart (cardiomegaly), osteoarthritis, spinal cord compression or fractures, increased risk of cancerous tumors, precancerous growths (polyps) on the lining of the colon.[4]
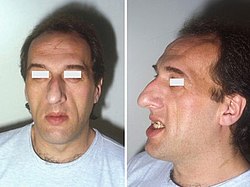
Acromegaly is a disorder that results in excess growth of certain parts of the human body. It is caused by excess growth hormone (GH) after the growth plates have closed. The initial symptom is typically enlargement of the hands and feet.[3] There may also be an enlargement of the forehead, jaw, and nose. Other symptoms may include joint pain, thickened skin, deepening of the voice, headaches, and problems with vision.[3] Complications of the disease may include type 2 diabetes, sleep apnea, and high blood pressure.[3]
Compared with the hand of an unaffected person (left), the hand of a person with acromegaly (right) is enlarged, with fingers that are widened, thickened and stubby, and with thicker soft tissue
Enlargement of the hands, feet, nose, lips, and ears, and a general thickening of the skin
Soft tissue swelling of internal organs, notably the heart with the attendant weakening of its muscularity, and the kidneys, also the vocal cords resulting in a characteristic thick, deep voice and slowing of speech
Generalized expansion of the skull at the fontanelle
Pronounced brow protrusion, often with ocular distension (frontal bossing)
Pronounced lower jaw protrusion (prognathism) with attendant macroglossia (enlargement of the tongue) and teeth spacing
Compression of the optic chiasm by the growth of pituitary adenoma leading to visual problems[13]
Causes
Pituitary adenoma
About 98% of cases of acromegaly are due to the overproduction of growth hormone by a benign tumor of the pituitary gland called an adenoma.[14] These tumors produce excessive growth hormone and compress surrounding brain tissues as they grow larger. In some cases, they may compress the optic nerves. Expansion of the tumor may cause headaches and visual disturbances. In addition, compression of the surrounding normal pituitary tissue can alter the production of other hormones, leading to changes in menstruation and breast discharge in women and impotence in men because of reduced testosterone production.[15]
A marked variation in rates of GH production and the aggressiveness of the tumor occurs. Some adenomas grow slowly and symptoms of GH excess are often not noticed for many years. Other adenomas grow rapidly and invade surrounding brain areas or the sinuses, which are located near the pituitary. In general, younger people tend to have more aggressive tumors.[16]
Most pituitary tumors arise spontaneously and are not genetically inherited. Many pituitary tumors arise from a genetic alteration in a single pituitary cell that leads to increased cell division and tumor formation. This genetic change, or mutation, is not present at birth but is acquired during life. The mutation occurs in a gene that regulates the transmission of chemical signals within pituitary cells; it permanently switches on the signal that tells the cell to divide and secrete growth hormones. The events within the cell that cause disordered pituitary cell growth and GH oversecretion currently are the subject of intensive research.[16]
Pituitary adenomas and diffuse somatomammotroph hyperplasia may result from somatic mutations activating GNAS, which may be acquired or associated with McCune–Albright syndrome.[17][18]
Other tumors
In a few people, acromegaly is caused by tumors of the pancreas, lungs, and adrenal glands. These tumors also lead to an excess of GH, either because they produce GH themselves or, more frequently, because they produce GHRH (growth hormone-releasing hormone), the hormone that stimulates the pituitary to make GH. When these nonpituitary tumors are surgically removed, GH levels fall and the symptoms of acromegaly improve.[citation needed]
Diagnosis
Frequent serum GH measurements in normal subjects (left panel) demonstrate that GH can fluctuate between undetectable levels most of the time interspersed with peaks of up to 30 μg/L (90 mIU/L); in acromegaly (right panel) GH hypersecretion is continuous with no undetectable levels.
Diagnosis is by measuring growth hormone after a person has consumed a glucose solution, or by measuring insulin-like growth factor I in the blood. After diagnosis, medical imaging of the pituitary is carried out to determine if an adenoma is present. If excess growth hormone is produced during childhood, the result is the condition gigantism rather than acromegaly, and it is characterized by excessive height.[3]
An MRI of the brain focusing on the sella turcica after gadolinium administration allows for clear delineation of the pituitary and the hypothalamus and the location of the tumor.[citation needed]
Differential diagnosis
Pseudoacromegaly is a condition with the usual acromegaloid features but without an increase in growth hormone and IGF-1. It is frequently associated with insulin resistance.[19] Cases have been reported due to minoxidil at an unusually high dose.[20] It can also be caused by a selective post-receptor defect of insulin signalling, leading to the impairment of metabolic, but preservation of mitogenic, signaling.[21]
Treatment options include surgery to remove the tumor, medications, and radiation therapy. Surgery is usually the preferred treatment; the smaller the tumor, the more likely surgery will be curative. If surgery is contraindicated or not curative, somatostatin analogues or GH receptor antagonists may be used. Radiation therapy may be used if neither surgery nor medications are completely effective.[3] Without treatment, life expectancy is reduced by 10 years; with treatment, life expectancy is not reduced.[6]
Medications
Somatostatin analogues
The primary medical treatment of acromegaly is to use somatostatin analogues – octreotide (Sandostatin) or lanreotide (Somatuline).
Somatostatin analogues are also sometimes used to shrink large tumors before surgery.[22]
Because octreotide inhibits gastrointestinal and pancreatic function, long-term use causes digestive problems such as loose stools, nausea, and gas in one-third of people. In addition, approximately 25 percent of people with acromegaly develop gallstones, which are usually asymptomatic.[23] In some cases, octreotide treatment can cause diabetes because somatostatin and its analogues can inhibit the release of insulin.[citation needed] With an aggressive adenoma that is not able to be operated on, there may be a resistance to octreotide in which case a second-generation SSA, pasireotide, may be used for tumor control. However, insulin and glucose levels should be carefully monitored as pasireotide has been associated with hyperglycemia by reducing insulin secretion.[24]
Dopamine agonists
For those who are unresponsive to somatostatin analogues, or for whom they are otherwise contraindicated, it is possible to treat using cabergoline. As tablets rather than injections, they cost considerably less. These drugs can also be used as an adjunct to somatostatin analogue therapy. They are most effective in those whose pituitary tumours also secrete prolactin. Side effects of these dopamine agonists include gastrointestinal upset, nausea, vomiting, light-headedness when standing, and nasal congestion. These side effects can be reduced or eliminated if medication is started at a very low dose at bedtime, taken with food, and gradually increased to the full therapeutic dose.[citation needed]
Growth hormone receptor antagonists
The latest development in the medical treatment of acromegaly is the use of growth hormone receptor antagonists. The only available member of this family is pegvisomant (Somavert). By blocking the action of the endogenous growth hormone molecules, this compound is able to control the disease activity of acromegaly in virtually everyone with acromegaly. Pegvisomant has to be administered subcutaneously by daily injections. Combinations of long-acting somatostatin analogues and weekly injections of pegvisomant seem to be equally effective as daily injections of pegvisomant.[citation needed]
Paltusotine (Palsonify) was approved for medical use in the United States in September 2025.[25]
Surgery
Surgical removal of the pituitary tumor is usually effective in lowering growth hormone levels. Two surgical procedures are available for use. The first is endonasal transsphenoidal surgery, which involves the surgeon reaching the pituitary through an incision in the nasal cavity wall. The wall is reached by passing through the nostrils with microsurgical instruments. The second method is transsphenoidal surgery during which an incision is made into the gum beneath the upper lip. Further incisions are made to cut through the septum to reach the nasal cavity, where the pituitary is located. Endonasal transsphenoidal surgery is a less invasive procedure with a shorter recovery time than the older method of transsphenoidal surgery, and the likelihood of removing the entire tumor is greater with reduced side effects. Consequently, endonasal transsphenoidal surgery is the more common surgical choice.[citation needed]
These procedures normally relieve the pressure on the surrounding brain regions and lead to a lowering of GH levels. Surgery is most successful in people with blood GH levels below 40ng/ml before the operation and with pituitary tumors no larger than 10mm in diameter. Success depends on the skill and experience of the surgeon. The success rate also depends on what level of GH is defined as a cure. The best measure of surgical success is the normalization of GH and IGF-1 levels. Ideally, GH should be less than 2ng/ml after an oral glucose load. Complications of surgery may include cerebrospinal fluid leaks, meningitis, or damage to the surrounding normal pituitary tissue, requiring lifelong pituitary hormone replacement.[citation needed]
Even when surgery is successful and hormone levels return to normal, people must be carefully monitored for years for possible recurrence. More commonly, hormone levels may improve, but not return completely to normal. These people may then require additional treatment, usually with medications.[citation needed]
Radiation therapy
Radiation therapy is usually reserved for people who have tumours remaining after surgery. These people often also receive medication to lower GH levels. Radiation therapy is given in divided doses over four to six weeks. This treatment lowers GH levels by about 50 percent over 2 to 5 years. People monitored for more than 5 years show significant further improvement. Radiation therapy causes a gradual loss of production of other pituitary hormones with time. Loss of vision and brain injury, which have been reported, are very rare complications of radiation treatments.[citation needed]
Prognosis
Life expectancy of people with acromegaly is dependent on how early the disease is detected.[26] Life expectancy after the successful treatment of early disease is equal to that of the general population.[27] Acromegaly can often go on for years before diagnosis, resulting in poorer outcome, and it is suggested that the better the growth hormone is controlled, the better the outcome.[26] Upon successful surgical treatment, headaches and visual symptoms tend to resolve.[9] One exception is sleep apnea, which is present in around 70% of cases but does not tend to resolve with successful treatment of growth hormone level.[8] While hypertension is a complication of 40% of cases, it typically responds well to regular regimens of blood pressure medication.[8] Diabetes that occurs with acromegaly is treated with the typical medications, but successful lowering of growth hormone levels often alleviates symptoms of diabetes.[8] Hypogonadism without gonad destruction is reversible with treatment.[8] Acromegaly is associated with a slightly elevated risk of cancer.[28]
Epidemiology, history, and culture
Acromegaly affects about 6 per 100,000 people. It is most commonly diagnosed in middle age.[3] Males and females are affected with equal frequency.[29] It was first described in the medical literature by Nicolas Saucerotte in 1772.[30][31] The term is from the Greekἄκρον (akron) meaning "extremity", and μέγα (mega) meaning "large".[3]
Paul Benedict (1938–2008), American actor. Best known for portraying Harry Bentley, The Jeffersons' English next door neighbour[32]
Mary Ann Bevan (1874–1933), an English woman, who after developing acromegaly, toured the sideshow circuit as "the ugliest woman in the world".[33]
Eddie Carmel, born Oded Ha-Carmeili (1936–1972), Mandatory Palestine-born entertainer with gigantism and acromegaly, popularly known as "The Jewish Giant".[citation needed]
Rondo Hatton (1894–1946), American journalist and actor. A Hollywood favorite in B-movie horror films of the 1930s and 1940s. Hatton's disfigurement, due to acromegaly, developed over time, beginning during his service in World War I.[34]
Irwin Keyes (1952–2015), American actor. Best known for portraying Hugo Mojoloweski, George's occasional bodyguard on The Jeffersons[35]
The Great Khali (Dalip Singh Rana), Indian professional wrestler, is best known for his tenure with WWE under the ring name The Great Khali. He had his pituitary tumor removed in 2012 at age 39.[39]
André the Giant (André Roussimoff, 1946–1993), French professional wrestler and actor, known for playing Fezzik in The Princess Bride.
The French Angel (Maurice Tillet, 1903–1954), Russian-born French professional wrestler, is better known by his ring name, the French Angel.[40]
Pío Pico, the last Mexican Governor of California (1801–1894), manifested acromegaly without gigantism between at least 1847 and 1858. Some time after 1858, signs of the growth hormone-producing tumor disappeared along with all the secondary effects the tumor had caused in him. He looked normal in his 90s.[41] His remarkable recovery is likely an example of spontaneous selective pituitary tumor apoplexy.[42]
A number of notable historical figures have been, moreover, argued to have had acromegaly, despite no strict diagnosis of the condition.
Maximinus Thrax, Roman emperor (c.173, reigned 235–238). Descriptions, as well as depictions, indicate acromegaly, though remains of his body are yet to be found.[citation needed]
Lorenzo de' Medici (1449–92) may have had acromegaly. Historical documents and portraits, as well as a later analysis of his skeleton, support the speculation.[53]
Pianist and composer Sergei Rachmaninoff (1873–1943), noted for his hands that could comfortably stretch a 13th on the piano, was never diagnosed with acromegaly in his lifetime, but a medical article from 2006 suggests that he might have had it.[54]
↑Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J (8 April 2015). Harrison's Principles of Internal Medicine (19thed.). McGraw Hill. pp.2269–2271. ISBN978-0-07-180215-4.
↑Yaqub, Abid; Yaqub, Nadia (1 September 2008). "Insulin-mediated pseudoacromegaly: a case report and review of the literature". West Virginia Medical Journal. 104 (5): 12–16. PMID18846753. GaleA201087184.
↑Nguyen KH, Marks JG (June 2003). "Pseudoacromegaly induced by the long-term use of minoxidil". Journal of the American Academy of Dermatology. 48 (6): 962–5. doi:10.1067/mjd.2003.325. PMID12789195.
↑Gutfreund, Hanoch and Jürgen Renn. The Formative Years of Relativity: The History and Meaning of Einstein's Princeton Lectures. Princeton University Press. 2017. Page 393.
↑Denis, Brian. Einstein: A Life. John Wiley & Sons. 1997. Page 169.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.