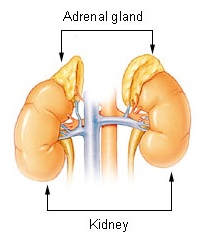
The adrenal glands are endocrine glands that produce a variety of hormones including adrenaline and the steroids aldosterone and cortisol. They are found above the kidneys. Each gland has an outer cortex which produces steroid hormones and an inner medulla. The adrenal cortex itself is divided into three main zones: the zona glomerulosa, the zona fasciculata and the zona reticularis.

Cushing's syndrome is a collection of signs and symptoms due to prolonged exposure to glucocorticoids such as cortisol. Signs and symptoms may include high blood pressure, abdominal obesity but with thin arms and legs, reddish stretch marks, a round red face due to facial plethora, a fat lump between the shoulders, weak muscles, weak bones, acne, and fragile skin that heals poorly. Women may have more hair and irregular menstruation. Occasionally there may be changes in mood, headaches, and a chronic feeling of tiredness.

Addison's disease, also known as primary adrenal insufficiency, is a rare long-term endocrine disorder characterized by inadequate production of the steroid hormones cortisol and aldosterone by the two outer layers of the cells of the adrenal glands, causing adrenal insufficiency. Symptoms generally come on slowly and insidiously and may include abdominal pain and gastrointestinal abnormalities, weakness, and weight loss. Darkening of the skin in certain areas may also occur. Under certain circumstances, an adrenal crisis may occur with low blood pressure, vomiting, lower back pain, and loss of consciousness. Mood changes may also occur. Rapid onset of symptoms indicates acute adrenal failure, which is a clinical emergency. An adrenal crisis can be triggered by stress, such as from an injury, surgery, or infection.
Cushing's disease is one cause of Cushing's syndrome characterised by increased secretion of adrenocorticotropic hormone (ACTH) from the anterior pituitary. This is most often as a result of a pituitary adenoma or due to excess production of hypothalamus CRH that stimulates the synthesis of cortisol by the adrenal glands. Pituitary adenomas are responsible for 80% of endogenous Cushing's syndrome, when excluding Cushing's syndrome from exogenously administered corticosteroids. The equine version of this disease is Pituitary pars intermedia dysfunction.
Adrenal insufficiency is a condition in which the adrenal glands do not produce adequate amounts of steroid hormones. The adrenal glands—also referred to as the adrenal cortex—normally secrete glucocorticoids, mineralocorticoids, and androgens. These hormones are important in regulating blood pressure, electrolytes, and metabolism as a whole. Deficiency of these hormones leads to symptoms ranging from abdominal pain, vomiting, muscle weakness and fatigue, low blood pressure, depression, mood and personality changes to organ failure and shock. Adrenal crisis may occur if a person having adrenal insufficiency experiences stresses, such as an accident, injury, surgery, or severe infection; this is a life-threatening medical condition resulting from severe deficiency of cortisol in the body. Death may quickly follow.

Hypopituitarism is the decreased (hypo) secretion of one or more of the eight hormones normally produced by the pituitary gland at the base of the brain. If there is decreased secretion of one specific pituitary hormone, the condition is known as selective hypopituitarism. If there is decreased secretion of most or all pituitary hormones, the term panhypopituitarism is used.

Sheehan's syndrome, also known as postpartum pituitary gland necrosis, occurs when the pituitary gland is damaged due to significant blood loss and hypovolemic shock usually during or after childbirth leading to decreased functioning of the pituitary gland (hypopituitarism). The pituitary gland is an endocrine organ, meaning it produces certain hormones and is involved in the regulation of various other hormones. This gland is located in the brain and sits in a pocket of the sphenoid bone known as the sella turcica. The pituitary gland works in conjunction with the hypothalamus, and other endocrine organs to modulate numerous bodily functions including growth, metabolism, menstruation, lactation, and even the "fight-or-flight" response. These endocrine organs release hormones in very specific pathways, known as hormonal axes. For example, the release of a hormone in the hypothalamus will target the pituitary to trigger the release of a subsequent hormone, and the pituitary's released hormone will target the next organ in the pathway. Hence, damage to the pituitary gland can have downstream effects on any of the aforementioned bodily functions.
Optic nerve hypoplasia (ONH) is a medical condition arising from the underdevelopment of the optic nerve(s). This condition is the most common congenital optic nerve anomaly. The optic disc appears abnormally small, because not all the optic nerve axons have developed properly. It is often associated with endocrinopathies, developmental delay, and brain malformations. The optic nerve, which is responsible for transmitting visual signals from the retina to the brain, has approximately 1.2 million nerve fibers in the average person. In those diagnosed with ONH, however, there are noticeably fewer nerves.

Pituitary adenomas are tumors that occur in the pituitary gland. Most pituitary tumors are benign, approximately 35% are invasive and just 0.1% to 0.2% are carcinomas. Pituitary adenomas represent from 10% to 25% of all intracranial neoplasms and the estimated prevalence rate in the general population is approximately 17%.

A prolactinoma is a tumor (adenoma) of the pituitary gland that produces the hormone prolactin. It is the most common type of functioning pituitary tumor. Symptoms of prolactinoma are due to abnormally high levels of prolactin in the blood (hyperprolactinemia), or due to pressure of the tumor on surrounding tissues. Based on size, a prolactinoma can be classified as a microprolactinoma or a macroprolactinoma.

Endocrine glands are ductless glands of the endocrine system that secrete their products, hormones, directly into the blood. The major glands of the endocrine system include the pineal gland, pituitary gland, pancreas, ovaries, testicles, thyroid gland, parathyroid gland, hypothalamus and adrenal glands. The hypothalamus and pituitary glands are neuroendocrine organs.
Nelson's syndrome is a disorder that occurs in about one in four patients who have had both adrenal glands removed to treat Cushing's disease. In patients with pre-existing adrenocorticotropic hormone (ACTH)-secreting pituitary adenomas, loss of adrenal feedback following bilateral adrenalectomy can trigger the rapid growth of the tumor, leading to visual symptoms and hyperpigmentation. The severity of the disease is dependent upon the effect of ACTH release on the skin, pituitary hormone loss from mass compression, as well as invasion into surrounding structures around the pituitary gland.

Adrenalectomy is the surgical removal of one or both adrenal glands. It is usually done to remove tumors of the adrenal glands that are producing excess hormones or is large in size. Adrenalectomy can also be done to remove a cancerous tumor of the adrenal glands, or cancer that has spread from another location, such as the kidney or lung. Adrenalectomy is not performed on those who have severe coagulopathy or whose heart and lungs are too weak to undergo surgery. The procedure can be performed using an open incision (laparotomy) or minimally invasive laparoscopic or robot-assisted techniques. Minimally invasive techniques are increasingly the gold standard of care due to shorter length of stay in the hospital, lower blood loss, and similar complication rates.

Adrenocorticotropic hormone deficiency is a rare disorder characterized by secondary adrenal insufficiency with minimal or no cortisol production and normal pituitary hormone secretion apart from ACTH. ACTH deficiency may be congenital or acquired, and its symptoms are clinically similar to those of glucocorticoid deficiency. Symptoms consist of weight loss, diminished appetite, muscle weakness, nausea, vomiting, and hypotension. Low blood sugar and hyponatremia are possible; however, blood potassium levels typically remain normal because affected patients are deficient in glucocorticoids rather than mineralocorticoids because of their intact renin-angiotensin-aldosterone system. ACTH may be undetectable in blood tests, and cortisol is abnormally low. Glucocorticoid replacement therapy is required. With the exception of stressful situations, some patients with mild or nearly asymptomatic disease may not require glucocorticoid replacement therapy. As of 2008 about two hundred cases have been described in the literature.
In humans and other animals, the adrenocortical hormones are hormones produced by the adrenal cortex, the outer region of the adrenal gland. These polycyclic steroid hormones have a variety of roles that are crucial for the body’s response to stress, and they also regulate other functions in the body. Threats to homeostasis, such as injury, chemical imbalances, infection, or psychological stress, can initiate a stress response. Examples of adrenocortical hormones that are involved in the stress response are aldosterone and cortisol. These hormones also function in regulating the conservation of water by the kidneys and glucose metabolism, respectively.
Autoimmune hypophysitis is defined as inflammation of the pituitary gland due to autoimmunity.

A pituitary disease is a disorder primarily affecting the pituitary gland.

Acromegaly is a disorder that results in excess growth of certain parts of the human body. It is caused by excess growth hormone (GH) after the growth plates have closed. The initial symptom is typically enlargement of the hands and feet. There may also be an enlargement of the forehead, jaw, and nose. Other symptoms may include joint pain, thicker skin, deepening of the voice, headaches, and problems with vision. Complications of the disease may include type 2 diabetes, sleep apnea, and high blood pressure.

Myelolipoma is a benign tumor-like lesion composed of mature adipose (fat) tissue and haematopoietic (blood-forming) elements in various proportions.
Galactorrhea hyperprolactinemia is increased blood prolactin levels associated with galactorrhea. It may be caused by such things as certain medications, pituitary disorders and thyroid disorders. The condition can occur in males as well as females. Relatively common etiologies include prolactinoma, medication effect, kidney failure, granulomatous diseases of the pituitary gland, and disorders which interfere with the hypothalamic inhibition of prolactin release. Ectopic (non-pituitary) production of prolactin may also occur. Galactorrhea hyperprolactinemia is listed as a “rare disease” by the Office of Rare Diseases of the National Institutes of Health. This means that it affects less than 200,000 people in the United States population.


