Hypopituitarism is the decreased (hypo) secretion of one or more of the eight hormones normally produced by the pituitary gland at the base of the brain.[1][2] If there is decreased secretion of one specific pituitary hormone, the condition is known as selective hypopituitarism.[3] If there is decreased secretion of most or all pituitary hormones, the term panhypopituitarism (pan meaning "all") is used.[4]
The signs and symptoms of hypopituitarism vary, depending on which hormones are under-secreted and on the underlying cause of the abnormality. The diagnosis of hypopituitarism is made by blood tests, but often specific scans and other investigations are needed to find the underlying cause, such as tumors of the pituitary, and the ideal treatment. Most hormones controlled by the secretions of the pituitary can be replaced by tablets or injections. Hypopituitarism is a rare disease, but may be significantly under-diagnosed in people with previous traumatic brain injury.[1] The first description of the condition was made in 1914 by the German physician Dr Morris Simmonds.[5]
Signs and symptoms
The hormones of the pituitary have different actions in the body, and the symptoms of hypopituitarism therefore depend on which hormone is deficient. The symptoms may be subtle and are often initially attributed to other causes.[1][6] In most of the cases, three or more hormones are deficient.[7] The most common problem is insufficiency of follicle-stimulating hormone (FSH) and/or luteinizing hormone (LH) leading to sex hormone abnormalities. Growth hormone deficiency is more common in people with an underlying tumor than those with other causes.[1][7]
Sometimes, there are additional symptoms that arise from the underlying cause; for instance, if the hypopituitarism is due to a growth hormone-producing tumor, there may be symptoms of acromegaly (enlargement of the hands and feet, coarse facial features), and if the tumor extends to the optic nerve or optic chiasm, there may be visual field defects. Headaches may also accompany pituitary tumors,[1] as well as pituitary apoplexy (infarction or haemorrhage of a pituitary tumor) and lymphocytic hypophysitis (autoimmune inflammation of the pituitary).[8] Apoplexy, in addition to sudden headaches and rapidly worsening visual loss, may also be associated with double vision that results from compression of the nerves in the adjacent cavernous sinus that control the eye muscles.[9]
Pituitary failure results in many changes in the skin, hair and nails as a result of the absence of pituitary hormone action on these sites.[10]
Complications
Several hormone deficiencies associated with hypopituitarism may lead to secondary diseases. For instance, growth hormone deficiency is associated with obesity, raised cholesterol and the metabolic syndrome, and estradiol deficiency may lead to osteoporosis. While effective treatment of the underlying hormone deficiencies may improve these risks, it is often necessary to treat them directly.[6]
Anterior pituitary
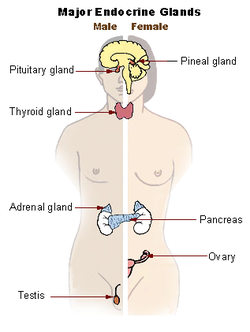
The major endocrine glands of the body. Pituitary hormones control the function of the adrenal gland, thyroid gland and the gonads (testes and ovaries).
Deficiency of all anterior pituitary hormones is more common than individual hormone deficiency.
Deficiency of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), together referred to as the gonadotropins, leads to different symptoms in men and women. Women experience oligo- or amenorrhea (infrequent/light or absent menstrual periods respectively) and infertility. Men lose facial, scrotal and trunk hair, as well as have decreased muscle mass and anemia. Both sexes may experience a decrease in libido and loss of sexual function, and have an increased risk of osteoporosis (bone fragility). Lack of LH/FSH in children is associated with delayed puberty.[1][6]
Prolactin (PRL) plays a role in breastfeeding, and inability to breastfeed may point at abnormally low prolactin levels.[8]
Posterior pituitary
Antidiuretic hormone (ADH) deficiency leads to the syndrome of diabetes insipidus (unrelated to diabetes mellitus): inability to concentrate the urine, leading to polyuria (production of large amounts of clear urine) that is low in solutes, dehydration and—in compensation—extreme thirst and constant need to drink (polydipsia), as well as hypernatremia (high sodium levels in the blood).[12] ADH deficiency may be masked if there is ACTH deficiency, with symptoms only appearing when cortisol has been replaced.[8]
Oxytocin (OXT) deficiency generally causes few symptoms,[1] however may lead to abnormal social developments due to its complex role as a social neuropeptide.[13]
Congenital hypopituitarism (present at birth) may be the result of complications around delivery, or may be the result of insufficient development (hypoplasia) of the gland, sometimes in the context of specific genetic abnormalities. Mutations may cause either insufficient development of the gland or decreased function. Forms of combined pituitary hormone deficiency ("CPHD") include:
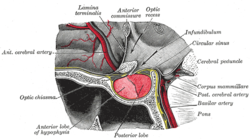
The pituitary gland is located at the base of the brain, and intimately connected with the hypothalamus. It consists of two lobes: the posterior pituitary, which consists of nervous tissue branching out of the hypothalamus, and the anterior pituitary, which consists of hormone-producing epithelium. The posterior pituitary secretes antidiuretic hormone, which regulates osmolarity of the blood, and oxytocin, which causes contractions of the uterus in childbirth and participates in breastfeeding.[14]
The pituitary develops in the third week of embryogenesis from interactions between the diencephalon part of the brain and the nasal cavity. The brain cells secrete FGF-8, Wnt5a and BMP-4, and the oral cavity BMP-2. Together, these cellular signals stimulate a group of cells from the oral cavity to form Rathke's pouch, which becomes independent of the nasal cavity and develops into the anterior pituitary; this process includes the suppression of production of a protein called Sonic hedgehog by the cells of Rathke's pouch.[16] The cells then differentiate further into the various hormone-producing cells of the pituitary. This requires particular transcription factors that induce the expression of particular genes. Some of these transcription factors have been found to be deficient in some forms of rare combined pituitary hormone deficiencies (CPHD) in childhood. These are HESX1, PROP1, POU1F1, LHX3, LHX4, TBX19, SOX2 and SOX3. Each transcription factor acts in particular groups of cells. Therefore, various genetic mutations are associated with specific hormone deficiencies.[16][17] For instance, POU1F1 (also known as Pit-1) mutations cause specific deficiencies in growth hormone, prolactin and TSH.[14][16][17] In addition to the pituitary, some of the transcription factors are also required for the development of other organs; some of these mutations are therefore also associated with specific birth defects.[16][17]
Most of the hormones in the anterior pituitary are each part of an axis that is regulated by the hypothalamus. The hypothalamus secretes a number of releasing hormones, often according to a circadian rhythm, into blood vessels that supply the anterior pituitary; most of these are stimulatory (thyrotropin-releasing hormone, corticotropin-releasing hormone, gonadotropin-releasing hormone and growth hormone-releasing hormone), apart from dopamine, which suppresses prolactin production.[18] In response to the releasing hormone rate, the anterior pituitary produces its hormones (TSH, ACTH, LH, FSH, GH) which in turn stimulate effector hormone glands in the body, while prolactin (PRL) acts directly on the breast gland. Once the effector glands produce sufficient hormones (thyroxine, cortisol, estradiol or testosterone and IGF-1), both the hypothalamus and the pituitary cells sense their abundance and reduce their secretion of stimulating hormones. The hormones of the posterior pituitary are produced in the hypothalamus and are carried by nerve endings to the posterior lobe; their feedback system is therefore located in the hypothalamus, but damage to the nerve endings would still lead to a deficiency in hormone release.[1]
Unless the pituitary damage is being caused by a tumor that overproduces a particular hormone, it is the lack of pituitary hormones that leads to the symptoms described above, and an excess of a particular hormone would indicate the presence of a tumor. The exception to this rule is prolactin: if a tumor compresses the pituitary stalk, a decreased blood supply means that the lactotrope cells, which produce prolactin, are not receiving dopamine and therefore produce excess prolactin. Hence, mild elevations in prolactin are attributed to stalk compression. Very high prolactin levels, though, point more strongly towards a prolactinoma (prolactin-secreting tumor).[6][19]
Diagnosis
CT scan of the brain showing a craniopharyngioma (white structure in the center of the image). This tumor may cause hypopituitarism and requires surgical removal.
The diagnosis of hypopituitarism is made on blood tests. Two types of blood tests are used to confirm the presence of a hormone deficiency: basal levels, where blood samples are taken–usually in the morning–without any form of stimulation, and dynamic tests, where blood tests are taken after the injection of a stimulating substance. Measurement of ACTH and growth hormone usually requires dynamic testing, whereas the other hormones (LH/FSH, prolactin, TSH) can typically be tested with basal levels. There is no adequate direct test for ADH levels, but ADH deficiency can be confirmed indirectly; oxytocin levels are not routinely measured.[1]
Generally, the finding of a combination of a low pituitary hormone together with a low hormone from the effector gland is indicative of hypopituitarism.[14] Occasionally, the pituitary hormone may be normal but the effector gland hormone decreased; in this case, the pituitary is not responding appropriately to effector hormone changes, and the combination of findings is still suggestive of hypopituitarism.[6]
Basal tests
Levels of LH/FSH may be suppressed by a raised prolactin level, and are therefore not interpretable unless prolactin is low or normal. In men, the combination of low LH and FSH in combination with a low testosterone confirms LH/FSH deficiency; a high testosterone would indicate a source elsewhere in the body (such as a testosterone-secreting tumor). In women, the diagnosis of LH/FSH deficiency depends on whether the woman has been through the menopause. Before the menopause, abnormal menstrual periods together with low estradiol and LH/FSH levels confirm a pituitary problem; after the menopause (when LH/FSH levels are normally elevated and the ovaries produce less estradiol), inappropriately low LH/FSH alone is sufficient.[1] Stimulation tests with GnRH are possible, but their use is not encouraged.[6][8]
For TSH, basal measurements are usually sufficient, as well as measurements of thyroxine to ensure that the pituitary is not simply suppressing TSH production in response to hyperthyroidism (an overactive thyroid gland). A stimulation test with thyrotropin-releasing hormone (TRH) is not regarded as useful.[8] Prolactin can be measured by basal level, and is required for the interpretation of LH and FSH results in addition to the confirmation of hypopituitarism or diagnosis of a prolactin-secreting tumor.[1]
Stimulation tests
Growth hormone deficiency is almost certain if all other pituitary tests are also abnormal, and insulin-like growth factor 1 (IGF-1) levels are decreased. If this is not the case, IGF-1 levels are poorly predictive of the presence of GH deficiency; stimulation testing with the insulin tolerance test is then required. This is performed by administering insulin to lower the blood sugar to a level below 2.2mmol/L. Once this occurs, growth hormone levels are measured. If they are low despite the stimulatory effect of the low blood sugars, growth hormone deficiency is confirmed. The test is not without risks, especially in those prone to seizures or are known to have heart disease, and causes the unpleasant symptoms of hypoglycemia.[1][6] Alternative tests (such as the growth hormone releasing hormone stimulation test) are less useful, although a stimulation test with arginine may be used for diagnosis, especially in situations where an insulin tolerance test is thought to be too dangerous.[20] If GH deficiency is suspected, and all other pituitary hormones are normal, two different stimulation tests are needed for confirmation.[8]
If morning cortisol levels are over 500nmol/L, ACTH deficiency is unlikely, whereas a level less than 100 is indicative. Levels between 100 and 500 require a stimulation test.[6] This, too, is done with the insulin tolerance test. A cortisol level above 500 after achieving a low blood sugar rules out ACTH deficiency, while lower levels confirm the diagnosis. A similar stimulation test using corticotropin-releasing hormone (CRH) is not sensitive enough for the purposes of the investigation. If the insulin tolerance test yields an abnormal result, a further test measuring the response of the adrenal glands to synthetic ACTH (the ACTH stimulation test) can be performed to confirm the diagnosis.[21] Stimulation testing with metyrapone is an alternative.[21] Some suggest that an ACTH stimulation test is sufficient as first-line investigation, and that an insulin tolerance test is only needed if the ACTH test is equivocal.[6][8] The insulin tolerance test is discouraged in children.[6] None of the tests for ACTH deficiency are perfect, and further tests after a period of time may be needed if initial results are not conclusive.[1]
Symptoms of diabetes insipidus should prompt a formal fluid deprivation test to assess the body's response to dehydration, which normally causes concentration of the urine and increasing osmolarity of the blood. If these parameters are unchanged, desmopressin (an ADH analogue) is administered. If the urine then becomes concentrated and the blood osmolarity falls, there is a lack of ADH due to lack of pituitary function ("cranial diabetes insipidus"). In contrast, there is no change if the kidneys are unresponsive to ADH due to a different problem ("nephrogenic diabetes insipidus").[1]
Further investigations
If one of these tests shows a deficiency of hormones produced by the pituitary, magnetic resonance imaging (MRI) scan of the pituitary is the first step in identifying an underlying cause. MRI may show various tumors and may assist in delineating other causes. Tumors smaller than 1cm are referred to as microadenomas, and larger lesions are called macroadenomas.[1]Computed tomography with radiocontrast may be used if MRI is not available.[8] Formal visual field testing by perimetry is recommended, as this would show evidence of optic nerve compression by a tumor.[8]
Treatment of hypopituitarism is threefold: removing the underlying cause, treating the hormone deficiencies, and addressing any other repercussions that arise from the hormone deficiencies.[1]
Pituitary tumors require treatment when they are causing specific symptoms, such as headaches, visual field defects or excessive hormone secretion. Transsphenoidal surgery (removal of the tumor by an operation through the nose and the sphenoidal sinuses) may, apart from addressing symptoms related to the tumor, also improve pituitary function, although the gland is sometimes damaged further as a result of the surgery. When the tumor is removed by craniotomy (opening the skull), recovery is less likely–but sometimes this is the only suitable way to approach the tumor.[1][19] After surgery, it may take some time for hormone levels to change significantly. Retesting the pituitary hormone levels is therefore performed 2 to 3 months later.[6]
Prolactinomas may respond to dopamine agonist treatment–medication that mimics the action of dopamine on the lactrotrope cells, usually bromocriptine or cabergoline. This approach may improve pituitary hormone secretion in more than half the cases, and make supplementary treatment unnecessary.[1][6][19][22]
Other specific underlying causes are treated as normally. For example, hemochromatosis is treated by venesection, the regular removal of a fixed amount of blood. Eventually, this decreases the iron levels in the body and improves the function of the organs in which iron has accumulated.[23]
Hormone replacement
Most pituitary hormones can be replaced indirectly by administering the products of the effector glands: hydrocortisone (cortisol) for adrenal insufficiency, levothyroxine for hypothyroidism, testosterone for male hypogonadism, and estradiol for female hypogonadism (usually with a progestogen to inhibit unwanted effects on the uterus). Growth hormone is available in synthetic form, but needs to be administered parenterally (by injection). Antidiuretic hormone can be replaced by desmopressin (DDAVP) tablets or nose spray. Generally, the lowest dose of the replacement medication is used to restore wellbeing and correct the deranged results, as excessive doses would cause side-effects or complications.[1][6][8] Those requiring hydrocortisone are usually instructed to increase their dose in physically stressful events such as injury, hospitalization and dental work as these are times when the normal supplementary dose may be inadequate, putting the patient at risk of adrenal crisis.[6][14]
Long-term follow up by specialists in endocrinology is generally needed for people with known hypopituitarism. Apart from ensuring the right treatment is being used and at the right doses, this also provides an opportunity to deal with new symptoms and to address complications of treatment.[6][8]
Difficult situations arise in deficiencies of the hypothalamus-pituitary-gonadal axis in people (both men and women) who experience infertility; infertility in hypopituitarism may be treated with subcutaneous infusions of FSH, human chorionic gonadotropin–which mimics the action of LH–and occasionally GnRH.[1][6][8]
Prognosis
Several studies have shown that hypopituitarism is associated with an increased risk of cardiovascular disease and some also an increased risk of death of about 50% to 150% the normal population.[6][14] It has been difficult to establish which hormone deficiency is responsible for this risk, as almost all patients studied had growth hormone deficiency.[8] The studies also do not answer the question as to whether the hypopituitarism itself causes the increased mortality, or whether some of the risk is to be attributed to the treatments, some of which (such as sex hormone supplementation) have a recognized adverse effect on cardiovascular risk.[8]
The largest study to date followed over a thousand people for eight years; it showed an 87% increased risk of death compared to the normal population. Predictors of higher risk were: female sex, absence of treatment for sex hormone deficiency, younger age at the time of diagnosis, and a diagnosis of craniopharyngioma. Apart from cardiovascular disease, this study also showed an increased risk of death from lung disease.[8][24]
Quality of life may be significantly reduced, even in those people on optimum medical therapy. Many report both physical and psychological problems. It is likely that the commonly used replacement therapies do not completely mimic the natural hormone levels in the body.[6] Health costs remain about double those of the normal population.[6]
Hypopituitarism is usually permanent. It requires lifelong treatment with one or more medicines.
Epidemiology
There is only one study that has measured the prevalence (total number of cases in a population) and incidence (annual number of new cases) of hypopituitarism.[1] This study was conducted in Northern Spain and used hospital records in a well-defined population. The study showed that 45.5 people out of 100,000 had been diagnosed with hypopituitarism, with 4.2 new cases per year.[7] 61% were due to tumors of the pituitary gland, 9% due to other types of lesions, and 19% due to other causes; in 11% no cause could be identified.[1][7]
Recent studies have shown that people with a previous traumatic brain injury, spontaneous subarachnoid hemorrhage (a type of stroke) or radiation therapy involving the head have a higher risk of hypopituitarism.[25] After traumatic brain injury, as much as a quarter have persistent pituitary hormone deficiencies.[26] Many of these people may have subtle or non-specific symptoms that are not linked to pituitary problems but attributed to their previous condition. It is therefore possible that many cases of hypopituitarism remain undiagnosed, and that the annual incidence would rise to 31 per 100,000 annually if people from these risk groups were to be tested.[1]
History
The pituitary was known to the ancients, such as Galen, and various theories were proposed about its role in the body, but major clues as to the actual function of the gland were not advanced until the late 19th century, when acromegaly due to pituitary tumors was described.[27] The first known report of hypopituitarism was made by the German physician and pathologist Dr Morris Simmonds. He described the condition on autopsy in a 46-year-old woman who had had severe puerperal fever eleven years earlier, and subsequently had amenorrhea, weakness, signs of rapid aging, and anemia. The pituitary gland was very small and there were few remnants of both the anterior and the posterior pituitary.[1][5] The eponym Simmonds' syndrome is used infrequently for acquired hypopituitarism, especially when cachexia (general ill health and malnutrition) predominates.[28][29] Most of the classic causes of hypopituitarism were described in the 20th century; the early 21st century saw the recognition of how common hypopituitarism could be in previous head injury victims.[1]
Until the 1950s, the diagnosis of pituitary disease remained based on clinical features and visual field examination, sometimes aided by pneumoencephalography and X-ray tomography. Nevertheless, the field of pituitary surgery developed during this time. The major breakthrough in diagnosis came with the discovery of radioimmunoassay by Rosalyn Yalow and Solomon Berson in the late 1950s.[30] This allowed the direct measurement of the hormones of the pituitary, which as a result of their low concentrations in blood had previously been hard to measure.[27] Stimulation tests were developed in the 1960s, and in 1973 the triple bolus test was introduced, a test that combined stimulation testing with insulin, GnRH and TRH. [31] Imaging of the pituitary, and therefore identification of tumors and other structural causes, improved radically with the introduction of computed tomography in the late 1970s and magnetic resonance imaging in the 1980s.[27]
1 2 3 4 Regal M, Páramo C, Sierra SM, Garcia-Mayor RV (December 2001). "Prevalence and incidence of hypopituitarism in an adult Caucasian population in northwestern Spain". Clin. Endocrinol. 55 (6): 735–40. doi:10.1046/j.1365-2265.2001.01406.x. PMID11895214. S2CID41502818.
↑ Pietrangelo A (June 2004). "Hereditary hemochromatosis--a new look at an old disease". N. Engl. J. Med. 350 (23): 2383–97. doi:10.1056/NEJMra031573. PMID15175440.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.