| Hamartoma | |
|---|---|
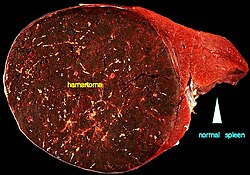 | |
| A large hamartoma of the spleen. The hamartoma is the dark circular object on the left that dominates the image. This is a cross-section; the growth is about 9 cm in diameter, while the entire spleen is about 11 cm in diameter. [1] | |
| Pronunciation |
|
| Specialty | Medical genetics, pathology |
| Diagnostic method | Chest x-ray, CT scan, MRI, ultrasound, and bronchoscopy. [2] |
A hamartoma is a mostly benign, [3] local malformation of cells that resembles a neoplasm of local tissue but is usually due to an overgrowth of multiple aberrant cells, with a basis in a systemic genetic condition, rather than a growth descended from a single mutated cell (monoclonality), as would typically define a benign neoplasm/tumor. [4] Despite this, many hamartomas are found to have clonal chromosomal aberrations that are acquired through somatic mutations, and on this basis the term hamartoma is sometimes considered synonymous with neoplasm. Hamartomas are by definition benign, slow-growing or self-limiting, [3] [4] though the underlying condition may still predispose the individual towards malignancies.
Contents
Hamartomas are usually caused by a genetic syndrome that affects the development cycle of all or at least multiple cells. [4] Many of these conditions are classified as overgrowth syndromes or cancer syndromes. Hamartomas occur in many different parts of the body and are most often asymptomatic incidentalomas (undetected until they are found incidentally on an imaging study obtained for another reason). Additionally, the definition of hamartoma versus benign neoplasm is often unclear, since both lesions can be clonal. Lesions such as adenomas, developmental cysts, hemangiomas, lymphangiomas and rhabdomyomas within the kidneys, lungs or pancreas are interpreted by some experts as hamartomas while others consider them true neoplasms. Moreover, even though hamartomas show a benign histology, there is a risk of some rare but life-threatening complications such as those found in neurofibromatosis type I and tuberous sclerosis. [5]
It is different from choristoma, a closely related form of heterotopia. [6] [7] The two can be differentiated as follows: a hamartoma is an excess of normal tissue in a normal situation (e.g., a birthmark on the skin), while a choristoma is an excess of tissue in an abnormal situation (e.g., pancreatic tissue in the duodenum). [8] [9] The term hamartoma is from the Greek ἁμαρτία, hamartia ("error"), and was introduced by Eugen Albrecht in 1904. [10]

