Calcium-activated chloride channel bestrophin-1 (BEST1), triple mutant: I76A, F80A, F84A; in complex with an Fab antibody fragment, chloride, and calcium. Secondary structure of biological assembly 1 viewed via front C5 axis orientation. From RCSB PDB.
The bestrophin family of proteins comprises four evolutionary related genes (BEST1, BEST2, BEST3, and BEST4) that code for integral membrane proteins.[6] This family was first identified in humans by linking a BEST1 mutation with Best vitelliform macular dystrophy (BVMD).[7] Mutations in the BEST1 gene have been identified as the primary cause for at least five different degenerative retinal diseases.[7]
The bestrophins are an ancient family of structurally conserved proteins that have been identified in nearly every organism studied from bacteria to humans. In humans, they function as calcium-activated anion channels, each of which has a unique tissue distribution throughout the body. Specifically, the BEST1 gene on chromosome 11q13 encodes the Bestrophin-1 protein in humans whose expression is highest in the retina.[7]
Structure
Gene
The bestrophin genes share a conserved gene structure, with almost identical sizes of the 8 RFP-TM domain-encoding exons and highly conserved exon-intron boundaries. Each of the four bestrophin genes has a unique 3-prime end of variable length.[5]
Calcium-activated chloride channel bestrophin-1 (BEST1), triple mutant: I76A, F80A, F84A; in complex with an Fab antibody fragment, chloride, and calcium. Subunit structure of Biological Assembly 1 viewed via side edge-centered orientation. From RCSB PDB
The structure of Best1 consists of five identical subunits that each span the membrane four times and form a continuous, funnel-shaped pore via the second transmembrane domain containing a high content of aromatic residues, including an invariant arg-phe-pro (RFP) motif.[7][11][12] The pore is lined with various nonpolar, hydrophobic amino acids. Both the structure and the composition of the pore help to ensure that only small anions are able to move completely through the channel. The channel acts as two funnels working together in tandem. It begins with a semi-selective, narrow entryway for anions, and then opens to a larger, positively charged area which then leads to a narrower pathway that further limits the size of anions passing through the pore. A calcium clasp acts as a belting mechanism around the larger, middle section of the channel. Calcium ions control the opening and closing of the channel due to conformational changes caused by calcium binding at the C-terminus directly following the last transmembrane domain.[7][12]
Tissue and subcellular distribution
The location of expression of the BEST1 gene is essential for protein functioning and mislocalization is often connected to a variety of retinaldegenerative diseases. The BEST1 gene expresses the Best1 protein primarily in the cytosol of the retinal pigment epithelium. The protein is typically contained in vesicles near the cellular membrane. There is also research to support that the Best1 protein is localized and produced in the endoplasmic reticulum (intracellular organelle involved in protein and lipid synthesis). Best1 is typically expressed with other proteins also synthesized in the endoplasmic reticulum, such as calreticulin, calnexin and Stim-1. Calcium ion involvement in the countertransport of chloride ions also supports the idea that Best1 is involved in forming calcium stores within the cell.[10]
Function
Best1 primarily functions as an intracellular calcium-activated chloride channel on the cellular membrane that is not voltage-dependent.[6][10][12] More recently Best1 has been shown to act as a volume-regulating anion channel.
Diseases
Best vitelliform macular dystrophy (BVMD)
Lipofuscin (lipid residual from lysosome digestion) in a human neuron. Representative of what may occur in the eye in people affected by BMVD.
Best's vitelliform macular dystrophy (BVMD) is one of the most common Best1-associated diseases. BVMD typically becomes noticeable in children and is represented by the buildup of lipofuscin (lipid residuals) lesions in the eye.[6][10] Diagnosis normally follows an abnormal electrooculogram in which decreased activation of calcium channels in the basolateral membrane of the retinal pigment epithelium becomes apparent. A mutation in the BEST1 gene leads to a loss of channel function and eventually retinal degeneration.[10] Although BVMD is an autosomal dominant form of macular dystrophy, expressivity varies within and between affected families although the overwhelming majority of affected families come from northern European descent.[7][10] Typically, people with this condition experience five progressively worsening stages, though timing and severity varies greatly. BVMD is often caused by the single missense mutations; however, amino acid deletions have also been identified.[7] A loss of function of the Best1 chloride channel could likely explain some of the most common issues associated with BVMD: an inability to regulate intracellular ion concentrations and regulate overall cell volume.[13] To date, over 100 disease-causing mutations have been related to BVMD as well as a number of other degenerative retinal diseases.[12] Although BVMD typically presents as a bilateral maculopathy, rare cases of persistent unilateral or markedly asymmetric BEST1-associated vitelliform disease have been reported, despite bilateral electrophysiological evidence of retinal pigment epithelium dysfunction.[14][15] In addition to genetic modifiers, a translational conceptual framework has been proposed in the context of asymmetric or unilateral expression in Best disease, termed Augmented Innate Silence (AIS). AIS defines “silence” as the absence of clinical disease expression despite the presence of genetic susceptibility. The concept of Augmented Innate Silence was proposed by Suneth Dayan Lindamulage and was inspired by clinical observations from the first reported case of unilateral Best disease from Sri Lanka.[15] AIS suggests that innate and locally acting protective mechanisms may suppress or delay disease manifestation in one eye. The framework of Augmented Innate Silence further proposes that identifying such mechanisms may inform future strategies aimed to augment this protective silence in disease-prone eyes, thereby guiding translational research into potential disease-modifying approaches.[16]
Adult-onset vitelliform macular dystrophy (AVMD)
Adult-onset vitelliform macular dystrophy (AVMD) consists of lesions similar to BVMD on the retina. However, the cause is not as definitive as BVMD. The inability to diagnosis AVMD via genetic testing makes differentiating between AVMD and pattern dystrophy difficult. It is also unknown whether there is truly a clinical difference between AVMD caused by BEST1 mutations and AVMD caused by PRPH2 mutations. AVMD usually involves less vision loss than BVMD and cases do not usually run in families.[7]
Autosomal recessive bestrophinopathy (ARB)
Autosomal recessive bestrophinopathy (ARB) was first identified in 2008. People with ARB demonstrate a decrease in vision during the first ten years of life. Parents and family members typically show no abnormalities as the disease is autosomal recessive, indicating that both alleles of the BEST1 gene must be mutated. Vitelliform lesions are often present and some cases involve cystoid macular edema. In addition, other complications have been observed. Vision decreases slowly over time, although rates of decline vary. Mutations causing ARB range from missense mutations to single base mutations in non-coding regions.[7]
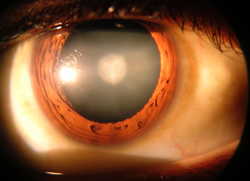
Cataract in human eye, potentially caused by autosomal dominant vitreoretinochoroidopathy.
Autosomal dominant vitreoretinochoroidopathy
Autosomal dominant vitreoretinochoroidopathy was first identified in 1982 and presents itself in both eyes with decreases in peripheral vision due to excessive fluid and changes in eye retinal pigmentation. Early onset cataracts are also likely.[7]
Retinitis pigmentosa (RP)
Fundus of patient with retinitis pigmentosa, mid stage
Retinitis pigmentosa was first described in relation to the BEST1 gene in 2009 and was found to be associated with four different missense mutations in the BEST1 gene in people. All affected individuals experience a diminished response to light within their retina and may have changes in pigmentation, pale optic discs, fluid accumulation and decreased visual acuity.[7]
All of the diseases above do not have any known treatments or cures. However, as of 2017, researchers are currently working on discovering treatments with stem cell transplants of the retinal pigment epithelium.[7]
1234567Strauss O, Neussert R, Müller C, Milenkovic VM (2012). "A potential cytosolic function of bestrophin-1". Retinal Degenerative Diseases. review. Advances in Experimental Medicine and Biology. Vol.723. pp.603–10. doi:10.1007/978-1-4614-0631-0_77. ISBN978-1-4614-0630-3. PMID22183384.
↑Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT (April 2008). "Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies". review. Physiological Reviews. 88 (2): 639–72. doi:10.1152/physrev.00022.2007. PMID18391176.
↑Olaf S, Müller C, Reichhart N, Tamm ER, Gomez NM (2014). "The Role of Bestrophin-1 in Intracellular Ca2+ Signaling". In Ash J, Grimm C, Hollyfield JG, Anderson RE, LaVail NM, Rickman CB (eds.). Retinal Degenerative Diseases: Mechanisms and Experimental Therapy. review. Advances in Experimental Medicine and Biology. Vol.801. New York: Springer. pp.113–119. doi:10.1007/978-1-4614-3209-8_15. ISBN978-1-4614-3209-8. PMID24664688.
↑Arora, R; etal. (2016). "Unilateral BEST1-Associated Retinopathy". American Journal of Ophthalmology. 169: 24–32. doi:10.1016/j.ajo.2016.05.024.
Forsman K, Graff C, Nordström S, Johansson K, Westermark E, Lundgren E, Gustavson KH, Wadelius C, Holmgren G (September 1992). "The gene for Best's macular dystrophy is located at 11q13 in a Swedish family". Clinical Genetics. 42 (3): 156–9. doi:10.1111/j.1399-0004.1992.tb03229.x. PMID1395087. S2CID924428.
Caldwell GM, Kakuk LE, Griesinger IB, Simpson SA, Nowak NJ, Small KW, Maumenee IH, Rosenfeld PJ, Sieving PA, Shows TB, Ayyagari R (May 1999). "Bestrophin gene mutations in patients with Best vitelliform macular dystrophy". Genomics. 58 (1): 98–101. doi:10.1006/geno.1999.5808. PMID10331951.
Bakall B, Marknell T, Ingvast S, Koisti MJ, Sandgren O, Li W, Bergen AA, Andreasson S, Rosenberg T, Petrukhin K, Wadelius C (May 1999). "The mutation spectrum of the bestrophin protein--functional implications". Human Genetics. 104 (5): 383–9. doi:10.1007/s004390050972. PMID10394929. S2CID21255716.
Allikmets R, Seddon JM, Bernstein PS, Hutchinson A, Atkinson A, Sharma S, Gerrard B, Li W, Metzker ML, Wadelius C, Caskey CT, Dean M, Petrukhin K (June 1999). "Evaluation of the Best disease gene in patients with age-related macular degeneration and other maculopathies". Human Genetics. 104 (6): 449–53. doi:10.1007/s004390050986. PMID10453731. S2CID6214287.
Palomba G, Rozzo C, Angius A, Pierrottet CO, Orzalesi N, Pirastu M (February 2000). "A novel spontaneous missense mutation in VMD2 gene is a cause of a best macular dystrophy sporadic case". American Journal of Ophthalmology. 129 (2): 260–2. doi:10.1016/S0002-9394(99)00327-X. PMID10682987.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.