Gene
The human CLCN5 gene (MIM#300008, reference sequence NG_007159.2) is localized in the pericentromeric region on chromosome Xp11.23. It extends over about 170 Kb of genomic DNA, has a coding region of 2,238 bp and consists of 17 exons including 11 coding exons (from 2 to 12). [5] [6] [7] [8] The CLCN5 gene has 8 paralogues ( CLCN1 , CLCN2 , CLCN3 , CLCN4, CLCN6 , CLCN7 , CLCNKA , CLCNKB ) and 201 orthologues among jawed vertebrates ( Gnathostomata ).
Five different CLCN5 gene transcripts have been discovered, two of which (transcript variants 3 [NM_000084.5] and 4 [NM_001282163.1]) encode for the canonical 746 amino acid protein, two (transcript variants 1 [NM_001127899.3] and 2 [NM_001127898.3]) for the NH2-terminal extended 816 amino acid protein [9] and one does not encode for any protein (Transcript variant 5, [NM_001272102.2]). The 5' untranslated region (5'UTR) of CLCN5 is complex and not entirely clarified. Two strong and one weak promoters were predicted to be present in the CLCN5 gene. [10] [11] Several different 5' alternatively used exons have been recognized in the human kidney. [9] [10] [11] [12] The three promoters drive with varying degree of efficiency 11 different mRNAs, with transcription initiating from at least three different start sites. [10]
Structure
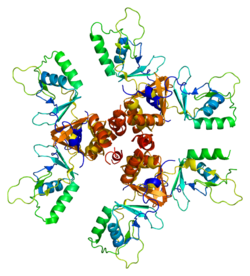
Like all ClC channels, ClC-5 needs to dimerize to create the pore through which the ions pass. [13] [14] [15] ClC-5 can form both homo- and hetero-dimers due to its marked sequence homology with ClC-3 and ClC-4. [16] [17] [18]
The canonical 746-amino acid ClC-5 protein has 18 membrane spanning α-helices (named A to R), an intracellular N- terminal domain and a cytoplasmic C-terminus containing two cystathionine beta-synthase (CBS) domains which are known to be involved in the regulation of ClC-5 activity. [13] [19] [20] [21] Helices B, H, I, O, P, and Q are the six major helices involved in the formation of dimer's interface and are crucial for proper pore configuration. [13] [14] The Cl− selectivity filter is principally driven by helices D, F, N, and R, which are conveyed together near the channel center. [13] [14] [22] [23] Two important amino acids for the proper ClC-5 function are the glutamic acids at position 211 and 268 called respectively "gating glutamate" and "proton glutamate". [24] [25] [26] [27] The gating glutamate is necessary for both H+ transport and ClC-5 voltage dependence. [8] [28] [29] The proton glutamate is crucial to the H+ transport acting as an H+ transfer site. [24] [30] [31]
Localization and function
ClC-5 belongs to the family of voltage gated chloride channel that are regulators of membrane excitability, transepithelial transport and cell volume in different tissues. Based on sequence homology, the nine mammalian ClC proteins can be grouped into three classes, of which the first (ClC-1, ClC-2, ClC-Ka and ClC-Kb) is expressed primarily in plasma membranes, whereas the other two (ClC-3, ClC-4, and ClC-5 and ClC-6 and ClC-7) are expressed primarily in organellar membranes. [32]
ClC-5 is expressed in minor to moderate level in brain, muscle, intestine but highly in the kidney, primarily in proximal tubular cells of S3 segment, in alfa intercalated cells of cortical collecting duct of and in cortical and medullary thick ascending limb of Henle's loop. [33] [34] [35] [36] [37] [38]
Proximal tubular cells (PTCs) are the main site of ClC-5 expression. By means of the receptor-mediated endocytosis process, they uptake albumin and low-molecular-weight proteins freely passed through the glomerular filter. ClC-5 is located in early endosomes of PTCs where it co-localizes with the electrogenic vacuolar H+-ATPase (V-ATPase). [34] [38] ClC-5 in this compartment contributes to the maintenance of intra-endosomal acidic pH. Environment acidification is necessary for the dissociation of ligand from its receptor. The receptor is then recycled to the apical membrane, while ligand is transported to the late endosome and lysosome where it is degraded. ClC-5 supports efficient acidification of endosomes either by providing a Cl− conductance to counterbalance the accumulation of positively charged H+ pumped in by V-ATPase or by directly acidifying endosome in parallel with V-ATPase. [39]
Experimental evidence indicates that endosomal Cl− concentration, which is raised by ClC-5 in exchange for protons accumulated by the V-ATPase, may play a role in endocytosis independently from endosomal acidification, thus pointing to another possible mechanism by which ClC-5 dysfunction may impair endocytosis. [40]
ClC-5 is located also at the cell surface of PTCs where probably it plays a role in the formation/function of the endocytic complex that also involves megalin and cubilin/amnionless receptors, the sodium-hydrogen antiporter 3 (NHE3), and the V-ATPase. [41] [42] It was demonstrated at the C-terminus of ClC-5 binds the actin-depolymerizing protein cofilin. When the nascent endosome forms, the recruitment of cofilin by ClC-5 is a prerequisite for the localized dissolution of the actin cytoskeleton, thus permitting the endosome to pass into the cytoplasm. It is conceivable that at the cell surface, the large intracellular C-terminus of ClC-5 has a crucial function in mediating the assembly, stabilization and disassembly of the endocytic complex via protein–protein interactions. Therefore, ClC-5 may accomplish two roles in the receptor-mediated endocytosis: i) vesicular acidification and receptor recycling; ii) participation to the non-selective megalin–cubilin-amnionless low-molecular-weight protein uptake at the apical membrane. [41]
Clinical significance
Dent disease is mainly caused by loss-of-function mutations in the CLCN5 gene (Dent disease 1; MIM#300009). [14] [43] Dent disease 1 shows a marked allelic heterogeneity. To date, 265 different CLCN5 pathogenic variants have been described. [14] A small number of pathogenic variants were found in more than one family. [44] The 48% are truncating mutations (nonsense, frameshift or complex), 37% non-truncating (missense or in-frame insertions/deletions), 10% splice site mutations, and 5% other type (large deletions, Alu insertions or 5'UTR mutations). Functional investigations in Xenopus laevis oocytes and mammalian cells [39] [43] [45] [46] [47] [40] enabled these CLCN5 mutations to be classified according to their functional consequences. [8] [44] [48] [49] [50] The most common mutations lead to a defective protein folding and processing, resulting in endoplasmic reticulum retention of the mutant protein for further degradation by the proteasome.
Animal models
Two independent ClC-5 knock-out mice, the so called Jentsch [51] [52] and Guggino models, [53] [54] [55] [56] provided critical insights into the mechanisms of proximal tubular dysfunction in Dent disease 1. These two murine models recapitulated the major features of Dent disease (low-molecular-weight proteinuria, hypercalciuria and nephrocalcinosis/nephrolithiasis) and demonstrated that ClC-5 inactivation is associated with severe impairment of both fluid phase and receptor-mediated endocytosis, as well as trafficking defects leading to the loss of megalin and cubilin at the brush border of proximal tubules. However, targeted disruption of ClC-5 in the Jentsch model did not lead to hypercalciuria, kidney stones or nephrocalcinosis, while the Guggino model did. [53] The Jentsch murine model produced slightly more acidic urines. Urinary phosphate excretion was increased in both models by about 50%. Hyperphosphaturia in the Jentsch model was associated with decreased apical expression of the sodium/phosphate cotransporter NaPi2a that is the predominant phosphate transporter in the proximal tubule. However, NaPi2a expression is ClC-5-independent since apical NaPi2a was normally expressed in any proximal tubules of chimeric female mice, while it was decreased in all male proximal tubular knock-out cells. Serum parathormone (PTH) is normal in knock-out mice while urinary PTH is increased of about 1.7 fold. Megalin usually mediates the endocytosis and degradation of PTH in proximal tubular cells. In knock-out mice, the downregulation of megalin leads to PTH defective endocytosis and progressively increases luminal PTH levels that enhance the internalization of NaPi2a. [51]
DNA testing and genetic counselling
A clinical diagnosis of Dent disease can be confirmed through molecular genetic testing that can detect mutations in specific genes known to cause Dent disease. However, about 20-25% of Dent disease patients remain genetically unresolved. [44]
Genetic testing is useful to determine the status of healthy carrier in the mother of an affected male. In fact, being Dent disease an X-linked recessive disorder, males are more frequently affected than females, and females may be heterozygous healthy carrier. Due to skewed X-inactivation, female carriers may present some mild symptoms of Dent disease such as low-molecular-weight proteinuria or hypercalciuria. Carriers will transmit the disease to half of their sons whereas half of their daughters will be carriers. Affected males do not transmit the disease to their sons since they pass Y chromosome to males, but all their daughters will inherited mutated X chromosome. Preimplant and prenatal genetic testing is not advised for Dent disease 1 since the prognosis for the majority of the patients is good and a clear correlation between genotype and phenotype is lacking. [57]
This page is based on this
Wikipedia article Text is available under the
CC BY-SA 4.0 license; additional terms may apply.
Images, videos and audio are available under their respective licenses.