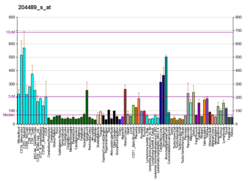
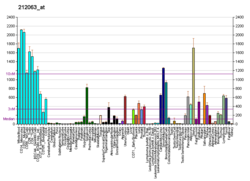
Kv7.1 (KvLQT1) is a potassiumchannelprotein whose primary subunit in humans is encoded by the KCNQ1gene.[5] Its mutation causes Long QT syndrome, Kv7.1 is a voltage and lipid-gated potassium channel present in the cell membranes of cardiac tissue and in inner ear neurons among other tissues. In the cardiac cells, Kv7.1 mediates the IKs (or slow delayed rectifying K+) current that contributes to the repolarization of the cell, terminating the cardiac action potential and thereby the heart's contraction. It is a member of the KCNQ family of potassium channels.
KvLQT1 is made of six membrane-spanning domains S1-S6, two intracellular domains, and a pore loop.[6] The KvLQT1 channel is made of four KCNQ1 subunits, which form the actual ion channel.
Function
This gene encodes a protein for a voltage-gated potassium channel required for the repolarization phase of the cardiac action potential. The gene product can form heteromultimers with two other potassium channel proteins, KCNE1 and KCNE3. The gene is located in a region of chromosome 11 that contains a large number of contiguous genes that are abnormally imprinted in cancer and the Beckwith-Wiedemann syndrome. Two alternative transcripts encoding distinct isoforms have been described.[7]
Clinical significance
Mutations in the gene can lead to a defective protein and several forms of inherited arrhythmias as Long QT syndrome[8] which is a prolongation of the QT interval of heart repolarization, Short QT syndrome,[8] and Familial Atrial Fibrillation. KvLQT1 are also expressed in the pancreas, and KvLQT1 Long QT syndrome patients has been shown to have hyperinsulinemic hypoglycaemia following an oral glucose load.[9] Currents arising from Kv7.1 in over-expression systems have never been recapitulated in native tissues - Kv7.1 is always found in native tissues with a modulatory subunit. In cardiac tissue, these subunits comprise KCNE1 and yotiao. Though physiologically irrelevant, homotetrameric Kv7.1 channels also display a unique form of C-type inactivation that reaches equilibrium quickly, allowing KvLQT1 currents to plateau. This is different from the inactivation seen in A-type currents, which causes rapid current decay.
KvLQT1 can also associate with any of the five members of the KCNE family of proteins, but interactions with KCNE1, KCNE2, KCNE3 are the only interactions within this protein family that affect the human heart. KCNE2, KCNE4, and KCNE5 have been shown to have an inhibitory effect on the functionality of KvLQT1, while KCNE1 and KCNE3 are activators of KvLQT1.[6] KvLQT1 can associate with KCNE1 and KCNE4 with the activation effects of KCNE1 overriding the inhibitory effects of KCNE4 on the KvLQT1 channel, and KvLQT1 will commonly associate with anywhere from two to four different KCNE proteins in order to be functional.[6] However, KvLQT1 most commonly associates with KCNE1 and forms the KvLQT1/KCNE1 complex since it has only been seen to function in vivo when associated with another protein.[6] KCNQ1 will form a heteromer with KCNE1 in order to slow its activation and enhance the current density at the plasma membrane of the neuron.[6][12] In addition to associating with KCNE proteins, the N-terminal juxtamembranous domain of KvLQT1 can also associate with SGK1, which stimulates the slow delayed potassium rectifier current. Since SGK1 requires structural integrity to stimulate KvLQT1/KCNE1, any mutations present in the KvLQT1 protein can result in reduced stimulation of this channel by SGK1.[13] General mutations in KvLQT1 have been known to cause a decrease in this slow delayed potassium rectifier current, longer cardiac action potentials, and a tendency to have tachyarrhythmias.[12]
KvLQT1/KCNE1
KCNE1 (minK), can assemble with KvLQT1 to form a slow delayed potassium rectifier channel. KCNE1 slows the inactivation of KvLQT1 when the two proteins form a heteromeric complex, and the current amplitude is greatly increased compared to WT-KvLQT1 homotetrameric channels. KCNE1 associates with the pore region of KvLQT1, and its transmembrane domain contributes to the selectivity filter of this heteromeric channel complex.[12] The alpha helix of the KCNE1 protein interacts with the pore domain S5/S6 and with the S4 domain of the KvLQT1 channel. This results in structural modifications of the voltage sensor and the selectivity filter of the KvLQT1 channel.[14] Mutations in either the alpha subunit of this complex, KvLQT1 or the beta subunit, KCNE1, can lead to Long QT Syndrome or other cardiac rhythmic deformities.[13] When associated with KCNE1, the KvLQT1 channel activates much more slowly and at a more positive membrane potential. It is believed that two KCNE1 proteins interact with a tetrameric KvLQT1 channel, since experimental data suggests that there are 4 alpha subunits and 2 beta subunits in this complex.[14] KVLQT1/KCNE1 channels are taken up from the plasma membrane through a RAB5 dependent mechanism, but inserted into the membrane by RAB11, a GTPase.[15]
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.