Waardenburg syndrome is a group of rare genetic conditions characterised by at least some degree of congenital hearing loss and pigmentation deficiencies, which can include bright blue eyes, a white forelock or patches of light skin. These basic features constitute type 2 of the condition; in type 1, there is also a wider gap between the inner corners of the eyes called telecanthus, or dystopia canthorum. In type 3, which is rare, the arms and hands are also malformed, with permanent finger contractures or fused fingers, while in type 4, the person also has Hirschsprung's disease. There also exist at least two types that can result in central nervous system (CNS) symptoms such as developmental delay and muscle tone abnormalities.

Otospondylomegaepiphyseal dysplasia (OSMED) is an autosomal recessive disorder of bone growth that results in skeletal abnormalities, severe hearing loss, and distinctive facial features. The name of the condition indicates that it affects hearing (oto-) and the bones of the spine (spondylo-), and enlarges the ends of bones (megaepiphyses).

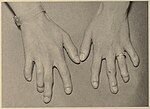
Popliteal pterygium syndrome (PPS) is an inherited condition affecting the face, limbs, and genitalia. The syndrome goes by a number of names including the popliteal web syndrome and, more inclusively, the facio-genito-popliteal syndrome. The term PPS was coined by Gorlin et al. in 1968 on the basis of the most unusual anomaly, the popliteal pterygium.
Adams–Oliver syndrome (AOS) is a rare congenital disorder characterized by defects of the scalp and cranium, transverse defects of the limbs, and mottling of the skin.

Laminopathies are a group of rare genetic disorders caused by mutations in genes encoding proteins of the nuclear lamina. Since the first reports of laminopathies in the late 1990s, increased research efforts have started to uncover the vital role of nuclear envelope proteins in cell and tissue integrity in animals. Laminopathies are a group of degenerative diseases, other disorders associated with inner nuclear membrane proteins are known as nuclear envelopathies.
CHIME syndrome, also known as Zunich–Kaye syndrome or Zunich neuroectodermal syndrome, is a rare congenital ichthyosis first described in 1983. The acronym CHIME is based on its main symptoms: colobomas, heart defects, ichthyosiform dermatosis, intellectual disability, and either ear defects or epilepsy. It is a congenital syndrome with only a few cases studied and published.
Vici syndrome, also called immunodeficiency with cleft lip/palate, cataract, hypopigmentation and absent corpus callosum, is a rare autosomal recessive congenital disorder characterized by albinism, agenesis of the corpus callosum, cataracts, cardiomyopathy, severe psychomotor retardation, seizures, immunodeficiency and recurrent severe infections. To date, about 50 cases have been reported.

Gap junction alpha-1 protein (GJA1), also known as connexin 43 (Cx43), is a protein that in humans is encoded by the GJA1 gene on chromosome 6. As a connexin, GJA1 is a component of gap junctions, which allow for gap junction intercellular communication (GJIC) between cells to regulate cell death, proliferation, and differentiation. As a result of its function, GJA1 is implicated in many biological processes, including muscle contraction, embryonic development, inflammation, and spermatogenesis, as well as diseases, including oculodentodigital dysplasia (ODDD), heart malformations, and cancers.

Gap junction beta-2 protein (GJB2), also known as connexin 26 (Cx26) — is a protein that in humans is encoded by the GJB2 gene.

Boomerang dysplasia is a lethal form of osteochondrodysplasia known for a characteristic congenital feature in which bones of the arms and legs are malformed into the shape of a boomerang. Death usually occurs in early infancy due to complications arising from overwhelming systemic bone malformations.

Frontonasal dysplasia (FND) is a congenital malformation of the midface. For the diagnosis of FND, a patient should present at least two of the following characteristics: hypertelorism, a wide nasal root, vertical midline cleft of the nose and/or upper lip, cleft of the wings of the nose, malformed nasal tip, encephalocele or V-shaped hair pattern on the forehead. The cause of FND remains unknown. FND seems to be sporadic (random) and multiple environmental factors are suggested as possible causes for the syndrome. However, in some families multiple cases of FND were reported, which suggests a genetic cause of FND.
EEM syndrome is an autosomal recessive congenital malformation disorder affecting tissues associated with the ectoderm, and also the hands, feet and eyes.

Fibrochondrogenesis is a rare autosomal recessive form of osteochondrodysplasia, causing abnormal fibrous development of cartilage and related tissues.
Gerodermia osteodysplastica (GO) is a rare autosomal recessive connective tissue disorder included in the spectrum of cutis laxa syndromes.
Scalp–ear–nipple (SEN) syndrome is a condition associated with aplasia cutis congenita.
Perlman syndrome (PS), also known as nephroblastomatosis-fetal ascites-macrosomia-Wilms tumor syndrome, is a rare overgrowth syndrome caused by autosomal recessive mutations in the DIS3L2 gene. PS is characterized by macrocephaly, neonatal macrosomia, nephromegaly, renal dysplasia, dysmorphic facial features, and increased risk for Wilms' tumor. The syndrome is associated with high neonatal mortality.

Gap junction gamma-2 (GJC2), also known as connexin-46.6 (Cx46.6) and connexin-47 (Cx47) and gap junction alpha-12 (GJA12), is a protein that in humans is encoded by the GJC2 gene.
Donnai–Barrow syndrome is a genetic disorder first described by Dian Donnai and Margaret Barrow in 1993. It is associated with LRP2. It is an inherited (genetic) disorder that affects many parts of the body.
Kenny-Caffey syndrome type 2 (KCS2) is an extremely rare autosomal dominant genetic condition characterized by dwarfism, hypermetropia, microphthalmia, and skeletal abnormalities. This subtype of Kenny-Caffey syndrome is caused by a heterozygous mutation in the FAM111A gene (615292) on chromosome 11q12.
Cole–Carpenter syndrome is an extremely rare autosomal recessive medical condition in humans. The condition affects less than 10 people worldwide. It is characterised by dysmorphic features and a tendency to fractures.