| Inborn error of steroid metabolism | |
|---|---|
 | |
| Steroidogenesis | |
| Specialty | Medical genetics, endocrinology |
An inborn error of steroid metabolism is an inborn error of metabolism due to defects in steroid metabolism.[ citation needed ]
| Inborn error of steroid metabolism | |
|---|---|
| | |
| Steroidogenesis | |
| Specialty | Medical genetics, endocrinology |
An inborn error of steroid metabolism is an inborn error of metabolism due to defects in steroid metabolism.[ citation needed ]
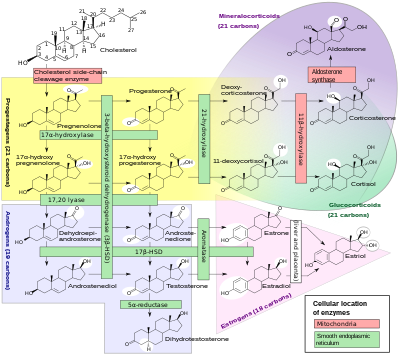
A variety of conditions of abnormal steroidogenesis exist due to genetic mutations in the steroidogenic enzymes involved in the process, of which include:
In addition, several conditions of abnormal steroidogenesis due to genetic mutations in receptors , as opposed to enzymes, also exist, including:[ citation needed ]
No activating mutations of the GnRH receptor in humans have been described in the medical literature, [3] and only one of the FSH receptor has been described, which presented as asymptomatic. [4] [5]

The adrenal glands are endocrine glands that produce a variety of hormones including adrenaline and the steroids aldosterone and cortisol. They are found above the kidneys. Each gland has an outer cortex which produces steroid hormones and an inner medulla. The adrenal cortex itself is divided into three main zones: the zona glomerulosa, the zona fasciculata and the zona reticularis.

Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders characterized by impaired cortisol synthesis. It results from the deficiency of one of the five enzymes required for the synthesis of cortisol in the adrenal cortex. Most of these disorders involve excessive or deficient production of hormones such as glucocorticoids, mineralocorticoids, or sex steroids, and can alter development of primary or secondary sex characteristics in some affected infants, children, or adults. It is one of the most common autosomal recessive disorders in humans.
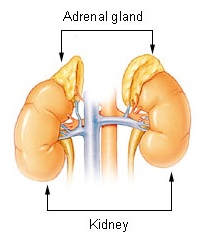
Adrenal insufficiency is a condition in which the adrenal glands do not produce adequate amounts of steroid hormones. The adrenal glands—also referred to as the adrenal cortex—normally secrete glucocorticoids, mineralocorticoids, and androgens. These hormones are important in regulating blood pressure, electrolytes, and metabolism as a whole. Deficiency of these hormones leads to symptoms ranging from abdominal pain, vomiting, muscle weakness and fatigue, low blood pressure, depression, mood and personality changes to organ failure and shock. Adrenal crisis may occur if a person having adrenal insufficiency experiences stresses, such as an accident, injury, surgery, or severe infection; this is a life-threatening medical condition resulting from severe deficiency of cortisol in the body. Death may quickly follow.

Lipoid congenital adrenal hyperplasia is an endocrine disorder that is an uncommon and potentially lethal form of congenital adrenal hyperplasia (CAH). It arises from defects in the earliest stages of steroid hormone synthesis: the transport of cholesterol into the mitochondria and the conversion of cholesterol to pregnenolone—the first step in the synthesis of all steroid hormones. Lipoid CAH causes mineralocorticoid deficiency in affected infants and children. Male infants are severely undervirilized causing their external genitalia to look feminine. The adrenals are large and filled with lipid globules derived from cholesterol.
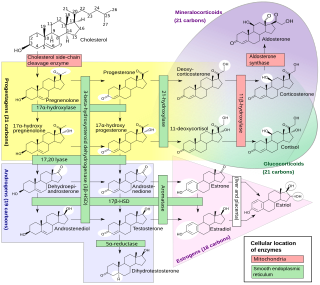
Congenital adrenal hyperplasia due to 11β-hydroxylase deficiency is a form of congenital adrenal hyperplasia (CAH) which produces a higher than normal amount of androgen, resulting from a defect in the gene encoding the enzyme steroid 11β-hydroxylase (11β-OH) which mediates the final step of cortisol synthesis in the adrenal. 11β-OH CAH results in hypertension due to excessive mineralocorticoid effects. It also causes excessive androgen production both before and after birth and can virilize a genetically female fetus or a child of either sex.

Congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase deficiency is an uncommon form of congenital adrenal hyperplasia (CAH) resulting from a mutation in the gene for one of the key enzymes in cortisol synthesis by the adrenal gland, 3β-hydroxysteroid dehydrogenase (3β-HSD) type II (HSD3B2). As a result, higher levels of 17α-hydroxypregnenolone appear in the blood with adrenocorticotropic hormone (ACTH) challenge, which stimulates adrenal corticosteroid synthesis.
Congenital adrenal hyperplasia due to 17α-hydroxylase deficiency is an uncommon form of congenital adrenal hyperplasia (CAH) resulting from a mutation in the gene CYP17A1, which produces the enzyme 17α-hydroxylase. It causes decreased synthesis of cortisol and sex hormones, with resulting increase in mineralocorticoid production. Thus, common symptoms include mild cortisol deficiency, ambiguous genitalia in men or amenorrhea at puberty in women, and hypokalemic hypertension. However, partial (incomplete) deficiency often has inconsistent symptoms between patients, and affected women may be asymptomatic except for infertility.
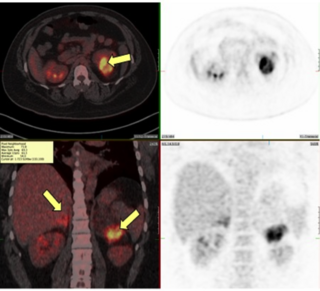
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency (CAH) is a genetic disorder characterized by impaired production of cortisol in the adrenal glands.

Cytochrome P450 17A1 is an enzyme of the hydroxylase type that in humans is encoded by the CYP17A1 gene on chromosome 10. It is ubiquitously expressed in many tissues and cell types, including the zona reticularis and zona fasciculata of the adrenal cortex as well as gonadal tissues. It has both 17α-hydroxylase and 17,20-lyase activities, and is a key enzyme in the steroidogenic pathway that produces progestins, mineralocorticoids, glucocorticoids, androgens, and estrogens. More specifically, the enzyme acts upon pregnenolone and progesterone to add a hydroxyl (-OH) group at carbon 17 position (C17) of the steroid D ring, or acts upon 17α-hydroxyprogesterone and 17α-hydroxypregnenolone to split the side-chain off the steroid nucleus.
Isolated 17,20-lyase deficiency (ILD), also called isolated 17,20-desmolase deficiency, is a rare endocrine and autosomal recessive genetic disorder which is characterized by a complete or partial loss of 17,20-lyase activity and, in turn, impaired production of the androgen and estrogen sex steroids. The condition manifests itself as pseudohermaphroditism in males, in whom it is considered to be a form of intersex, and, in both sexes, as a reduced or absent puberty/lack of development of secondary sexual characteristics, resulting in a somewhat childlike appearance in adulthood.
Leydig cell hypoplasia (LCH), also known as Leydig cell agenesis, is a rare autosomal recessive genetic and endocrine syndrome affecting an estimated 1 in 1,000,000 individuals with XY chromosomes. It is characterized by an inability of the body to respond to luteinizing hormone (LH), a gonadotropin which is normally responsible for signaling Leydig cells of the testicles to produce testosterone and other androgen sex hormones. The condition manifests itself as pseudohermaphroditism, hypergonadotropic hypogonadism, reduced or absent puberty, and infertility.
Follicle-stimulating hormone (FSH) insensitivity, or ovarian insensitivity to FSH in females, also referable to as ovarian follicle hypoplasia or granulosa cell hypoplasia in females, is a rare autosomal recessive genetic and endocrine syndrome affecting both females and males, with the former presenting with much greater severity of symptomatology. It is characterized by a resistance or complete insensitivity to the effects of follicle-stimulating hormone (FSH), a gonadotropin which is normally responsible for the stimulation of estrogen production by the ovaries in females and maintenance of fertility in both sexes. The condition manifests itself as hypergonadotropic hypogonadism, reduced or absent puberty, amenorrhea, and infertility in females, whereas males present merely with varying degrees of infertility and associated symptoms.

Seviteronel is an experimental cancer medication which is under development by Viamet Pharmaceuticals and Innocrin Pharmaceuticals for the treatment of prostate cancer and breast cancer. It is a nonsteroidal CYP17A1 inhibitor and works by inhibiting the production of androgens and estrogens in the body. As of July 2017, seviteronel is in phase II clinical trials for both prostate cancer and breast cancer. In January 2016, it was designated fast-track status by the United States Food and Drug Administration for prostate cancer. In April 2017, seviteronel received fast-track designation for breast cancer as well.
A steroidogenesis inhibitor, also known as a steroid biosynthesis inhibitor, is a type of drug which inhibits one or more of the enzymes that are involved in the process of steroidogenesis, the biosynthesis of endogenous steroids and steroid hormones. They may inhibit the production of cholesterol and other sterols, sex steroids such as androgens, estrogens, and progestogens, corticosteroids such as glucocorticoids and mineralocorticoids, and neurosteroids. They are used in the treatment of a variety of medical conditions that depend on endogenous steroids.

A CYP17A1 inhibitor is a type of drug that inhibits the enzyme CYP17A1. CYP17A1 inhibitors work by blocking specific enzyme functions, impacting androgen biosynthesis.
Adrenal steroids are steroids that are derived from the adrenal glands. They include corticosteroids, which consist of glucocorticoids like cortisol and mineralocorticoids like aldosterone, adrenal androgens like dehydroepiandrosterone (DHEA), DHEA sulfate (DHEA-S), and androstenedione (A4), and neurosteroids like DHEA and DHEA-S, as well as pregnenolone and pregnenolone sulfate (P5-S). Adrenal steroids are specifically produced in the adrenal cortex.
Walter L. Miller is an American endocrinologist and professor emeritus of pediatrics at the University of California, San Francisco (UCSF). Miller is expert in the field of human steroid biosynthesis and disorders of steroid metabolism. Over the past 40 years Miller's group at UCSF has described molecular basis of several metabolic disorders including, congenital adrenal hyperplasia, pseudo vitamin D dependent rickets, severe, recessive form of Ehlers-Danlos syndrome, 17,20 lyase deficiency caused by CYP17A1 defects, P450scc deficiency caused by CYP11A1 defects, P450 oxidoreductase deficiency.
Steroidogenic enzymes are enzymes that are involved in steroidogenesis and steroid biosynthesis. They are responsible for the biosynthesis of the steroid hormones, including sex steroids and corticosteroids, as well as neurosteroids, from cholesterol. Steroidogenic enzymes are most highly expressed in classical steroidogenic tissues, such as the testis, ovary, and adrenal cortex, but are also present in other tissues in the body.
An estrogen synthesis inhibitor is a type of drug which inhibits the enzymatic synthesis of estrogens, such as estradiol. They include:
Late onset congenital adrenal hyperplasia (LOCAH), also known as nonclassic congenital adrenal hyperplasia, is a milder form of congenital adrenal hyperplasia (CAH), a group of autosomal recessive disorders characterized by impaired cortisol synthesis that leads to variable degrees of postnatal androgen excess.